

Abstract

Potent imidazopyridine-based inhibitors of fatty acid synthase (FASN) are described. The compounds are shown to have antiviral (HCV replicon) activities that track with their biochemical activities. The most potent analogue (compound 19) also inhibits rat FASN and inhibits de novo palmitate synthesis in vitro (cell-based) as well as in vivo.
Keywords: FASN, inhibitor, antiviral, HCV, structure−activity relationship, in vivo, target modulation, reversible
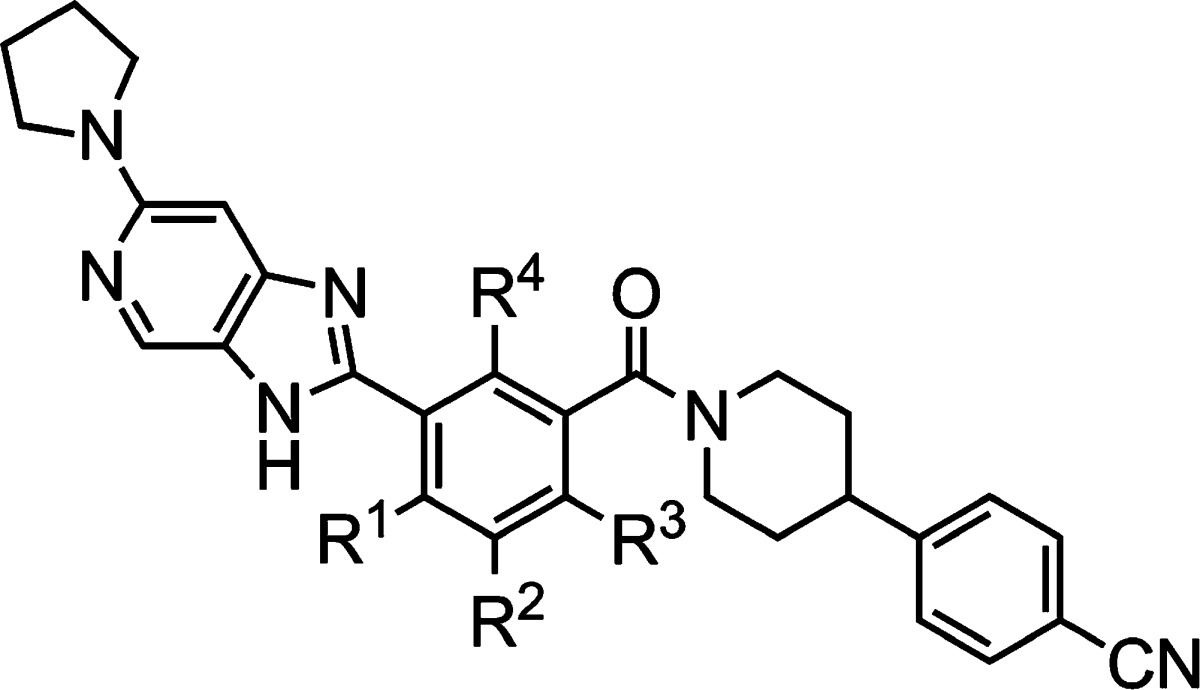
Human fatty acid synthase (hFASN) is a key enzyme in the de novo synthesis of fatty acids (FAs) including palmitate.1,1b While the putative involvement of FASN in cancer has been well documented,2−2c other potential therapeutic uses of FASN inhibitors are beginning to emerge including cardiovascular disease and diabetes.3−3c Furthermore, it has recently been shown that FASN is up-regulated during hepatitis C virus (HCV) infection and that reduction of FASN levels using siRNA leads to diminished replication of the virus.4 Infection by HCV has become a serious health issue with a significant portion (∼3%) of the world's population affected.5,5b While the majority of the newer HCV treatments are direct-acting antivirals (DAAs) that target viral proteins,5a it has been proposed that blocking the infectious cycle by modulating cellular factors such as FASN may result in a higher barrier to the emergence of resistance.6,6b
Although a number of inhibitors of FASN have been reported, the majority of them have had limited utility due to modest inhibitory activity or low cell permeability or were irreversible binders; the latter include the widely used tool compounds C75 and Cerulenin (Chart 1).2c,7−7c The goal of the current work was two-fold. First, we wished to identify potent, small-molecule inhibitors of FASN with a reversible mode of inhibition. Second, to enable studies in cell-based assays, good cell permeability was essential. Compounds with these characteristics were expected to be useful tools for evaluating the potential of FASN as a small-molecule drug target for the treatment of HCV.8 Furthermore, to pave the way for subsequent drug discovery efforts, the compounds needed to have good activity against both human and rodent FASN, along with reasonable rodent pharmacokinetic (PK) profiles, enabling pharmacodynamic (PD) studies.9
Chart 1. Inhibitors of FASN.

In spite of having very limited existing structure–activity relationships (SAR) or published data, compound 3 (Chart 1) and its close analogue 4 were among the more promising starting points at the inception of our explorations.7,10 However, while these molecules were potent biochemical inhibitors of human FASN (hFASN, Scheme 1), members of this class were found to undergo cleavage of the secondary amide moiety in rodent plasma resulting in the formation of a potentially toxic primary aniline. Furthermore, the weak activities of 3 and 4 against rat FASN (rFASN) precluded their use in preclinical in vivo studies. A search for modified structures, devoid of these issues, was therefore initiated (Scheme 1).
Scheme 1. Compound Progression Leading to Imidazopyridine Motif.

In an attempt to prevent amide cleavage, the parity of the amide was initially reversed, resulting in an equally active hFASN compound, 5 (Scheme 1), with good plasma stability (t1/2 > 10 h). However, the activity against rFASN was poor, and initial attempts to optimize this new series met with limited success. Hence, additional scaffold modifications were explored (Scheme 1). Cyclizing the reversed amide (5) yielded the corresponding imidazopyridine derivative 6. The new derivative, devoid of the secondary amide motif, had good plasma stability (t1/2 > 10 h). It also displayed promising biochemical activities against both hFASN and rFASN. The scaffold was therefore selected for further profiling and optimization. Compound 6 had good antiviral activity in a cell-based HCV replicon assay with a minor shift from the biochemical assay and an acceptable selectivity index (SI = EC50/CC50, where CC50 is cell viability EC50, Table 1). These observations are consistent with good cell permeability as confirmed in an Madin–Darby canine kidney (MDCK) permeability assay (Papp = 8.0 × 10–6 cm/s, efflux ratio = 2.5). Next, a methyl-walk around the central ring was conducted (Table 1). Replacing the R1-methyl with a hydrogen resulted in modest loss of human and rodent activities (compound 6 vs 7), whereas a methyl at R2 (compound 8) completely abolished biochemical and antiviral activities. On the other hand, replacing the R3-hydrogen with a methyl yielded an analogue (9) that had good activity against hFASN and also improved activity against the rodent enzyme. Finally, introducing a methyl at R4 again yielded a compound (10) devoid of activity against hFASN and rFASN. Overall, as shown previously, the antiviral activities track with biochemical inhibition of hFASN.8,11
Table 1. Preliminary SAR around Compound 6: Methyl-Walk, Antiviral, and FASN Inhibitory Activities.
| compd | R1 | R2 | R3 | R4 | hFASNa (IC50, μM) | rFASNb (IC50, μM) | HCVc (EC50, μM) | CC50d (IC50, μM) | SIe |
|---|---|---|---|---|---|---|---|---|---|
| 6 | Me | H | H | H | 0.107 | 1.72 | 0.175 | >20 | >114 |
| 7 | H | H | H | H | 0.130 | 2.95 | 0.550 | >3.0 | >5.0 |
| 8 | H | Me | H | H | >50 | >50 | >10 | >10 | N/A |
| 9 | H | H | Me | H | 0.060 | 0.635 | 0.150 | 8.6 | 57 |
| 10 | H | H | H | Me | >50 | >50 | >10 | 1.9 | <0.19 |
Biochemical inhibition of human FASN.
Biochemical inhibition of rat FASN.
Inhibition of HCV RNA in the replicon system. Telaprevir was used as a positive control.
Inhibition of cell viability.
SI = EC50/CC50; n ≥ 2, and CV ≤ 50%.
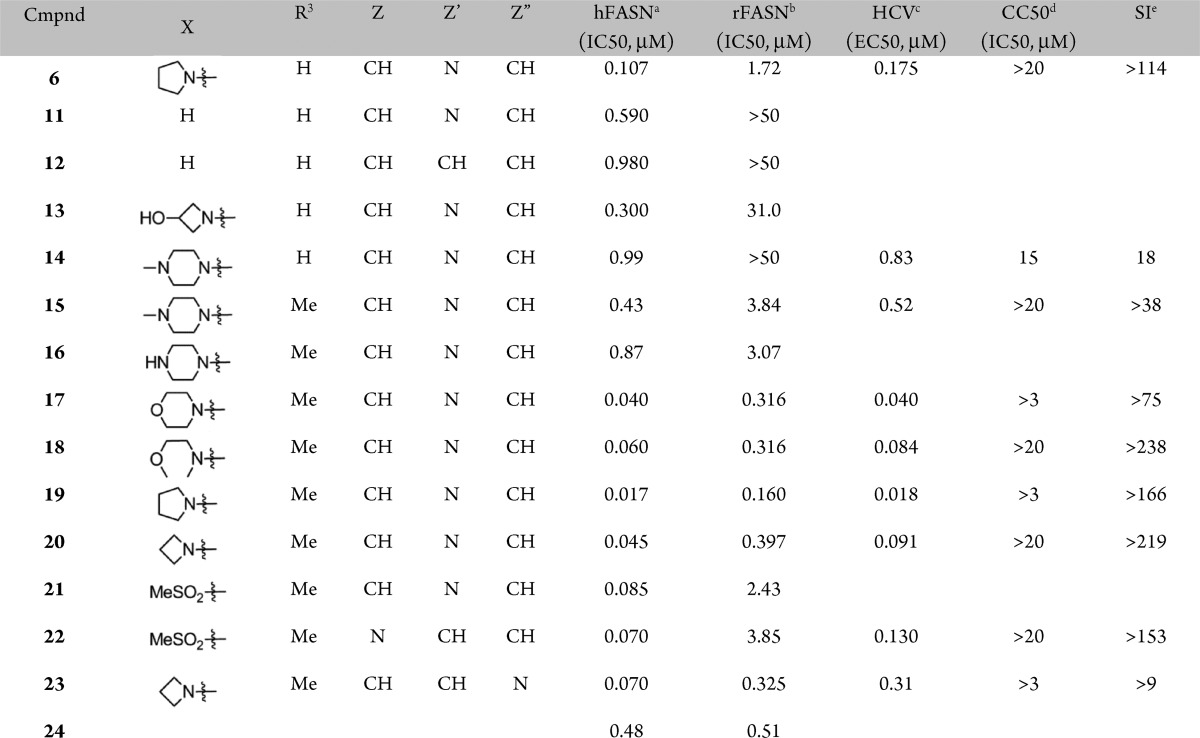
A preliminary survey of the imidazopyridine portion was undertaken next (Table 2). While replacing the pyrrolidine moiety in 6 with a hydrogen resulted in modest loss in potency against hFASN, a more significant decrease in activity was observed against the rat enzyme (compound 6 vs 11, Table 2). The corresponding unsubstituted benzimidazole analogue 12 showed further loss of activity.
Table 2. Imidazopyridine SAR, Antiviral, and FASN Inhibitory Activities.
Biochemical inhibition of human FASN.
Biochemical inhibition of rat FASN.
Inhibition of HCV RNA in the replicon system, typically determined only for more active compounds. See the Supporting Information for correlations across a larger number of analogues. Telaprevir was used as a positive control.
Inhibition of cell viability.
SI = EC50/CC50; n ≥ 2, and CV ≤ 50%.
Replacing the pyrrolidine moiety in 6 with polar substituents (compounds 13 and 14) rendered more soluble analogues but with a concomitant loss of rodent activity. In an attempt to regain some activity against rFASN (see above, compound 9, Table 1), the R3-methyl was introduced to yield piperazine derivative 15, which indeed diplayed improved activity against the rodent enzyme as compared to its R3 = hydrogen counterpart, compound 14. Therefore, subsequent analogues were prepared with the R3 = Me substitution pattern. While the corresponding unsubstituted piperazine analogue 16 did not show any improvement in activity, the noncharged morpholine and methyl ether derivatives (17 and 18) displayed good activities across the assays, including inhibition of rFASN and antiviral activity. Combining the original pyrrolidine moiety with the R3 = Me substitution pattern (compound 19) resulted in potent inhibition of hFASN and HCV replication. The activity against the rodent enzyme was also improved. While the corresponding azetidine derivative (20) showed a modest loss of activity, introduction of a methyl-sulfone in place of the pyrrolidne led to a more significant decrease, in particular against rFASN. Consistent with the SAR shown above, changing the position of the nitrogen in the imidazopyridine moiety resulted in minor changes in activities (compounds 20 vs 23 and 21 vs 22), whereas the effect of moving the X substituent is more pronounced (compound 20 vs 24). As noted above and previously, the cell-based antiviral activities are found to track well with the human biochemical activities.11 Further characterization using dialysis and washout studies indicated that compounds from this series are reversible inhibitors of FASN, both biochemically as well as in cell-based assays.8,9
The analogues described above were prepared using a general route exemplified by the preparation of compound 19 in Scheme 2. Treatment of 2,4-dimethylbenzoic acid (25) with iodine and sodium periodate in a mixture of sulfuric acid and acetic acid furnished the iodinated aryl compound 26 in 82% yield. Lithium-halogen exchange followed by addition of N,N-dimethylformamide (DMF) gave the corresponding aldehyde derivative (27) in good yield (74%). Amide coupling with 4-(piperidin-4-yl)benzonitrile12 was then achieved using HBTU in DMF to afford intermediate 28 (84%). Subsequent imidazole formation was accomplished by reacting aldehyde 28 with diamino-pyridine derivative 29 under oxidative conditions to give target compound 19 in 73% yield.
Scheme 2. Preparation of Compound 19.

Reagents and conditions: (a) NaIO4, I2, H2SO4, AcOH. (b) n-BuLi, THF, −78 °C, then DMF. (c) 4-(Piperidine-4-yl)benzonitrile, HBTU, DIEA, DMF. (d) Pyrrolidine, K2CO3, MeCN, 70 °C (91%). (e) H2, Pd/C, MeOH (94%). (f) Compound 28, Na2S2O5, DMF, 100 °C.
In the proton NMR spectrum of 19, the signal for the R3-methyl was split (Δδ ≈ 40 Hz, d6-DMSO), suggesting a possible hindered rotation around the phenyl-carbonyl bond. Preliminary estimates of rotational barriers in compound 19 were determined using VT-NMR.13,13b At a moderately elevated temperature (Tc < 323 K), the R3-methyl peaks coalesce, suggesting a fast interconversion between rotamers (<0.01 s at 37 °C).13−14d In a similar analysis of the rotation around the carbonyl-nitrogen bond, the coalescence temperature of piperidine protons (Tc ≈ 357 K, Δδ ≈ 65 Hz, d6-DMSO) implies a rotamer half-life of ∼0.1 s at 37 °C. Taken together, these preliminary observations suggest that 19 does not represent a mixture of discrete atropisomers.
Having identified potent inhibitors of human and rat FASN, we next studied the relationship between exposure and target modulation in vivo using 19 as a sentinel compound. De novo synthesis of palmitate was selected as a PD marker since palmitate is directly produced by FASN.1,9 As expected, 19 was a potent inhibitor of palmitate synthesis in both rat and human cells (Table 3). While compound 19 had moderate iv clearance and a short half-life in the rat (Table 4), its oral bioavailability and exposure over time were considered acceptable for an initial assessment of in vivo target modulation. The in vivo PD response was assessed in the liver since hepatocytes are the site of HCV infection.5b Livers were harvested 8 h after treating animals with oral doses of compound 19 delivered as a methylcellulose suspension (15 mg/kg, 50 mg/kg, or vehicle only), and the levels of compound and newly synthesized palmitate were determined. As shown in Table 3, the extent of palmitate synthesis inhibition 8 h after dosing is dose- and exposure-dependent. Strong inhibition of hepatic palmitate synthesis is observed at the higher dose (50 mg/kg), and a weaker response occurs at 15 mg/kg. The data in Table 3 indicate that the degree of suppression is correlated to the level of compound in the liver.
Table 3. In Vitro and in Vivo Inhibition of de Novo Palmitate Synthesis by Compound 19.
| human IC50 (HeLa, μM) | rat IC50 (NMU, μM) |
|---|---|
| in vitro | |
| 0.012 | 0.062 |
| rat no. | [19]liverb (μM) | % inhibitionc |
|---|---|---|
| in vivo: 15 mg/kga | ||
| 1 | 1.7 | 33 |
| 2 | 0.16 | 2 |
| 3 | 0.13 | 9 |
| 4 | 2.9 | 35 |
| 5 | 8.1 | 70 |
| in vivo: 50 mg/kga | ||
| 1 | 23.6 | 91 |
| 2 | 14.4 | 96 |
| 3 | 12.6 | 52 |
| 4 | 9.8 | 100 |
| 5 | 3.4 | 40 |
Oral dose.
Liver level of 19 at 8 h.
% inhibition of de novo palmitate synthesis at 8 h as compared to vehicle group.
Table 4. Rat PK Profile of Compound 19.
1 mg/kg.
50 mg/kg.
Clearance.
Volume of distribution.
Half-life.
Area under the curve.
Oral bioavailability.
In summary, the present study outlines the discovery of a potent FASN inhibitor that blocks palmitate synthesis in the liver. While compound 19 has desirable attributes including good cell permeability (MDCK Papp = 8.3 × 10–6 cm/s) and notable antiviral activity, its highly lipophilic nature (clogP = 4.99, cLipE = 2.75)15−15c and relatively high molecular weight (Mw = 504.6 g/mol, LE = 15.21)16,16b pose challenges to further development.17 Subsequent manuscripts will present our efforts to improve these properties while maintaining desired biological activities using the results and SAR presented in the current study as a foundation.
Acknowledgments
Excellent productivity and chemistry support from Dr. Tongqian Chen and Xiaojuan Hu and their team at Pharmaron is gratefully acknowledged.
Glossary
Abbreviations
- BuLi
butyl lithium
- DIEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide
- FASN
fatty acid synthase
- HCV
hepatitis C virus
- MDCK
Madin–Darby canine kidney
- PD
pharmacodynamic
- PK
pharmacokinetic
- SAR
structure–activity relationships
- SI
selectivity index
Supporting Information Available
Biological assays and experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): All authors are current or former employees of 3-V Biosciences, Inc..
Supplementary Material
References
- Chirala S. S.; Wakil S. J. Structure and function of animal fatty acid synthase. Lipids 2004, 39111045–1053. [DOI] [PubMed] [Google Scholar]
- Liu H.; Liu J. Y.; Wu X.; Zhang J. T. Biochemistry, molecular biology, and pharmacology of fatty acid synthase, an emerging therapeutic target and diagnosis/prognosis marker. Int. J. Biochem. Mol. Biol. 2010, 1169–89. [PMC free article] [PubMed] [Google Scholar]
- Kuhajda F. P. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition 2000, 163202–208. [DOI] [PubMed] [Google Scholar]
- Menendez J. A.; Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 710763–777. [DOI] [PubMed] [Google Scholar]
- Abramson H. N. The lipogenesis pathway as a cancer target. J. Med. Chem. 2011, 54165615–5638. [DOI] [PubMed] [Google Scholar]
- Menendez J. A.; Vazquez-Martin A.; Ortega F. J.; Fernandez-Real J. M. Fatty acid synthase: Association with insulin resistance, type 2 diabetes, and cancer. Clin. Chem. 2009, 553425–38. [DOI] [PubMed] [Google Scholar]
- Wakil S. J.; Abu-Elheiga L. A. Fatty acid metabolism: target for metabolic syndrome. J. Lipid Res. 2009, 50Suppl.S138–S143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.; Singh S. B.; Wang J.; Chung C. C.; Salituro G.; Karanam B. V.; Lee S. H.; Powles M.; Ellsworth K. P.; Lassman M. E.; Miller C.; Myers R. W.; Tota M. R.; Zhang B. B.; Li C. Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes. Proc. Natl. Acad. Sci. U.S.A. 2011, 108135378–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Hood B. L.; Chadwick S. L.; Liu S.; Watkins S. C.; Luo G.; Conrads T. P.; Wang T. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 2008, 4851396–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikova I. Hepatitis C—Pipeline update. Nat. Rev. Drug Discovery 2011, 10293–94. [DOI] [PubMed] [Google Scholar]
- Lemon S. M.; Walker M. C.; Alter M. J.; Yi M.. Hepatitis C Virus. In Fields Virology; Knipe D. M., Howley P. M., Griffin D. E., Lamb R. A., Martin M. A., Roizman B., Straus S. E., Eds.; Lippincott Williams & Wilkins: Philadelphia, 2006; Vol. 1, pp 1253–1304. [Google Scholar]
- Prussia A.; Thepchatri P.; Snyder J. P.; Plemper R. K. Systematic Approaches towards the Development of Host-Directed Antiviral Therapeutics. Int. J. Mol. Sci. 2011, 1264027–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwegmann A.; Brombacher F. Host-directed drug targeting of factors hijacked by pathogens. Sci. Signal 2008, 129re8. [DOI] [PubMed] [Google Scholar]
- Flavin R.; Peluso S.; Nguyen P. L.; Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010, 64551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- When tested in our biochemical assays, C75 and Cerulenin had weak activities (>5 μM) against human and rat fatty acid synthase.
- For a recent example of a novel inhibitor scaffold, see Abdel-Magid A. F.Fatty Acid Synthase Inhibitors as Possible Treatment for Cancer. ACS Med. Chem. Lett. 2012, 3 (8), 612–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble G.; McDowell R.; Moese S.; Oslob J. D.; Patick A. K.; Yendluri S.; Parsey M.. Potent Hepatitis C Antiviral Activity by Inhibiting Fatty Acid Synthase. The 63rd Annual Meeting of the American Association for the Study of Liver Diseases, Boston, MA, 2012. [Google Scholar]
- Evanchik M.; Cai H.; Feng Q. S.; Hu L.; Johnson R.; Kemble G.; Kosaka Y.; Lai J.; Oslob J. D.; Sivaraja M.; Tep S.; Yang H.; Zaharia C. A.; McDowell R.. TVB-2640, a Novel Anti-HCV Agent, Safely Causes Sustained Host-Target Inhibition In Vivo. The 63rd Annual Meeting of the American Association for the Study of Liver Diseases, Boston, MA, 2012. [Google Scholar]
- Wallenius L.; Butlin R.; Kjellstedt A.; Lofgren L.; Oakes N.. A Novel Fatty Acid Synthase Inhibitor Suppresses De Novo Lipogenesis but induces Hepatic Steatosis, Dermatitis and does not enhance Insulin Sensitivity in Obese Zucker Rats. American Diabetes Association 68th Scientific Sessions, San Francisco, CA, 2008. [Google Scholar]
- See Graph S1 in the Supporting Information.
- Pieper H.; Linz G.; Himmelsbach F.; Austel V.; Muller T.; Weisenberger J.; Guth B.. Carboxylic acid derivatives, pharmaceutical compositions containing these compounds and processes for preparing them. U.S. 5442064, 1995.
- Sandström J.Dynamic NMR Spectroscopy; Academic Press: London, New York, 1982. [Google Scholar]
- Friebolin H., Basic One- and Two-Dimensional NMR Spectroscopy; Wiley-VCH: Weinheim, New York, 1998. [Google Scholar]
- Clayden J.; Moran W. J.; Edwards P. J.; LaPlante S. R. The challenge of atropisomerism in drug discovery. Angew. Chem., Int. Ed. Engl. 2009, 48356398–6401. [DOI] [PubMed] [Google Scholar]
- Guile S. D.; Bantick J. R.; Cooper M. E.; Donald D. K.; Eyssade C.; Ingall A. H.; Lewis R. J.; Martin B. P.; Mohammed R. T.; Potter T. J.; Reynolds R. H.; St-Gallay S. A.; Wright A. D. Optimization of monocarboxylate transporter 1 blockers through analysis and modulation of atropisomer interconversion properties. J. Med. Chem. 2007, 502254–263. [DOI] [PubMed] [Google Scholar]
- LaPlante S. R.; Edwards P. J.; Fader L. D.; Jakalian A.; Hucke O. Revealing atropisomer axial chirality in drug discovery. ChemMedChem 2011, 63505–513. [DOI] [PubMed] [Google Scholar]
- Laplante S. R.; Fader L. D.; Fandrick K. R.; Fandrick D. R.; Hucke O.; Kemper R.; Miller S. P.; Edwards P. J. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 2011, 54207005–7022. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 611881–90. [DOI] [PubMed] [Google Scholar]
- Ryckmans T.; Edwards M. P.; Horne V. A.; Correia A. M.; Owen D. R.; Thompson L. R.; Tran I.; Tutt M. F.; Young T. Rapid assessment of a novel series of selective CB(2) agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 2009, 19154406–4409. [DOI] [PubMed] [Google Scholar]
- Here, cLipE = p(HCV EC50) – clogP.
- Perola E. An analysis of the binding efficiencies of drugs and their leads in successful drug discovery programs. J. Med. Chem. 2010, 5372986–2997. [DOI] [PubMed] [Google Scholar]
- Here, LE = p(hFAS IC50)/Mw × 1000.
- Edwards M. P.; Price D. A.. Role of Physicochemical Properties and Ligand Lipophilicity Efficiency in Addressing Drug Safety Risks. In Annual Reports in Medicinal Chemistry; Macor J. E., Ed.; Academic Press: Amsterdam, Boston, 2010; Vol. 45, Chapter 23, pp 380–391. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.