

Abstract
Fibroblast activation protein (FAP) is a serine protease that is generally accepted to play an important role in tumor growth and other diseases involving tissue remodeling. Currently there are no FAP inhibitors with reported selectivity toward both the closely related dipeptidyl peptidases (DPPs) and prolyl oligopeptidase (PREP). We present the discovery of a new class of FAP inhibitors with a N-(4-quinolinoyl)-Gly-(2-cyanopyrrolidine) scaffold. We have explored the effects of substituting the quinoline ring and varying the position of its sp2 hybridized nitrogen atom. The most promising inhibitors combined low nanomolar FAP inhibition and high selectivity indices (>103) with respect to both the DPPs and PREP. Preliminary experiments on a representative inhibitor demonstrate that plasma stability, kinetic solubility, and log D of this class of compounds can be expected to be satisfactory.
Keywords: Fibroblast activation protein (FAP), dipeptidyl peptidase IV (DPPIV), prolyl oligopeptidase (PREP), seprase
Fibroblast activation protein (FAP, FAP-α, seprase) is a type II transmembrane serine protease, belonging to the prolyl oligopeptidase family. This family comprises serine proteases that cleave peptides preferentially after proline residues. Other important members of this family that are expressed in the human proteome are prolyl oligopeptidase (PREP) and the dipeptidyl peptidases (DPPs): DPPIV, DPPII, and DPP8/9.1
FAP expression is seen on activated stromal fibroblasts and pericytes of 90% of common human epithelial tumors examined.2,3 It has been established that FAP expression promotes tumorigenesis in mouse models and that FAP inhibition can attenuate tumor growth.4,5 FAP is also highly expressed in lesions characterized by activated stromal tissue such as present in cirrhosis, fibrotic diseases, osteoarthritis and rheumatoid arthritis, keloidosis, and in healing wounds.6−11
FAP has been demonstrated to possess both dipeptidyl peptidase and endopeptidase activity, catalyzed by the same active site. Several studies have identified in vitro substrates for FAP. Peptides found to be substrates of FAP’s dipeptidyl peptidase activity include Neuropeptide Y, B-type natriuretic peptide, substance P, and peptide YY. Analogously, α2-antiplasmin, type I collagen, and gelatin were found to behave as in vitro substrates of the endopeptidase activity of FAP.12−14 Nonetheless, the relevance of these findings under in vivo conditions has to be confirmed.
Currently no inhibitors with low nanomolar FAP affinity and selectivity toward both PREP and the DPPs have been reported. Most research effort in the domain of FAP-inhibitor discovery to date has been centered around pyrrolidine-2-boronic acid derivatives.15,16 These compounds in general also display significant affinity for one or several DPPs.15,16 The most representative of this class, Val-boroPro (Talabostat, PT-100) 1 has reached phase II clinical trials (Table 1). It was evaluated as a therapeutic drug for, among others, metastatic kidney cancer, pancreatic adenocarcinoma, nonsmall cell lung cancer, and chronic lymphocytary leukemia (Figure 1). While talabostat in several of these trials was able to induce clinical response, safety concerns potentially related to the drug’s lack of selectivity led to its withdrawal from further development.17,18
Table 1. IC50s for Reference Compounds 1 and 2.
| IC50 (μM) |
|||||
|---|---|---|---|---|---|
| Nr | FAP | DPP IV | DPP9 | DPP II | PREP |
| 1 | 0.07 ± 0.01 | 0.022 ± 0.001 | NDa | 0.086 ± 0.007 | 0.98 ± 0.06 |
| 2 | 0.37 ± 0.002 | 0.0020 ± 0.0002 | >100 | >100 | >100 |
Not determined.
Figure 1.
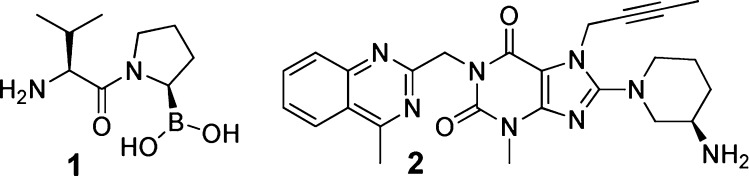
Structure of reference inhibitors used in this study: Val-boroPro (1) and linagliptin (2).
More recently, we and the group of Jiaang have focused on 2-cyanopyrrolidine derivatives for FAP inhibitor discovery.19,20 The 2-cyanopyrrolidine fragment has been successfully explored before for discovery of DPP inhibitors, exemplified, for example, by the marketed DPP IV inhibitors vildagliptin and saxagliptin.21−24 The work presented here deals with a group of 2-cyanopyrrolidines that represent the first examples of inhibitors combining low nanomolar FAP affinity and significant selectivity with respect to both PREP and the DPPs. Reference inhibitors used in this study are 1 and the FDA-approved, clinically used DPP IV inhibitor linagliptin (Tradjenta) 2 (Table 1).25 The latter compound is structurally distinct from 1 but has also been described to possess significant FAP affinity.
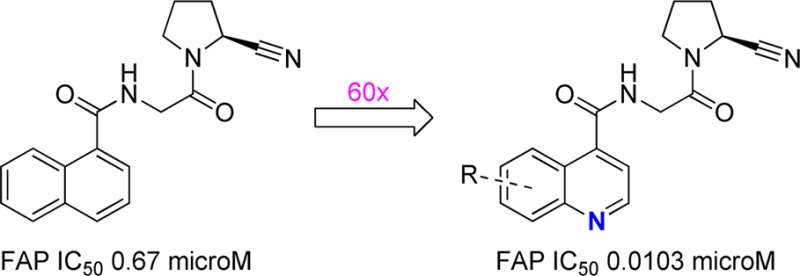
Previous results pointed out that the N-acyl-glycyl-(2-cyanopyrrolidine) scaffold has significant potential to deliver FAP inhibitors with good selectivity toward PREP and the dipeptidyl peptidases.19 The most potent compound identified (N-(1-naphthoyl)-Gly-(2-cyanopyrrolidine), 3) displayed high nanomolar FAP-affinity.19,20 For further optimization of this compound’s potency, we proposed further exploration of the P3 area, containing the naphtyl group. A docking study furthermore indicated that the 1-naphthoyl residue could be involved in a cation−π interaction with the guanidine side chain of FAP’s Arg123. A set of analogues potentially capable of corroborating this hypothesis and providing additional SAR information in the parts of chemical space surrounding the 1-naphthoyl group were prepared.
A general pathway for the synthesis of the target compounds 3–39 is displayed in Scheme 1. Commercially available prolinamide was coupled to N-Boc-glycine giving N-Boc-glycine-prolinamide 4 and subsequently dehydrated with trifluoroacetic anhydride to yield the corresponding nitrile 5. The nitrile was deprotected in acetonitrile to give hydrochloric acid salt 6, which was then coupled with a carboxylic acid or acyl chloride to give final compounds.
Scheme 1. Synthesis of Target Compounds 3-39.

Reagents and conditions: (a) Boc-glycine, HATU, DIPEA, DMF-DCM, rt, 71%; (b) trifluoroacetic anhydride, pyridine, THF, 0 °C, 92%; (c) (i) TFA, MeCN, 0 °C, 24h; (ii) HCl, diethylether, 84%; (d) HATU, DIPEA, R-COOH or 1-chloro-N,N,2-trimethylprop-1-en-1-amine, RCOOH, TEA or RCOCl, TEA, 20–76%.
All compounds synthesized in this study were evaluated as inhibitors of FAP, DPP IV, DPP9, DPP II, and PREP using a chromogenic substrate assay.19 It is worthwhile to stipulate that DPP9 potencies reported can reasonably be expected to also be indicative for inhibitor affinities toward the highly homologous DPP8.26,27
The results summarized in Table 2 show that the N-(4-quinolinoyl) substituted compound 7 has about 60 times more FAP-affinity than the initial N-(1-naphthoyl) based ‘hit’ 3. Its potency as an inhibitor of FAP also clearly stands out among the other compounds in Table 2. The presence of other azaheteroaromatic substituents, as, for example, in the N-(indolyl-3-acyl) containing 8 and its regio-isomeric congener 9, leads to a drastic drop in FAP-affinity. The same affinity trend was observed for compounds 10–12: in these molecules, the electron density in the naphthyl ring can reasonably be expected to be higher than in compound 3. Directly inspired by the docking study, these molecules were designed to gain affinity from a stronger cation−π interaction with FAP’s Arg123. Further deviation from ‘hit’ structure 3 was investigated with compounds 13 and 14 in which the acylated glycine amine function was changed for an oxygen atom. This, however, was also found to be detrimental for FAP potency. Likewise and in line with results obtained earlier, introduction of a sulfonamide function replacing the P3 acyl group in 15 decreases potency very significantly. The ((5-cyanopyridin-2-yl)amino)acetamide 16 and 3-hydroxyadamantane-1-carboxamide 17 are nonbasic analogues of the N-substituted glycyl-(2-cyanopyrroldine) DPPIV inhibitors NVP-DPP 728 and vildagliptin, respectively.24 By introducing an amide group instead of the parent structures’ basic P2-amine function that is critical for DPP IV interaction, we hoped to decrease DPP IV potency, while retaining affinity for the phylogenetically very closely related FAP. Only moderate inhibition of the latter was observed however. Finally, an analogue of compound 18 with a P1-pyrrolidine-2-boronate residue instead of a 2-cyanopyrrolidine group has been described as a low nanomolar FAP inhibitor.28 Nitrile 18, however, loses almost all FAP inhibition when compared to its boronate analogue.
Table 2. Enzymatic Evaluation Data for Compounds 3–18.
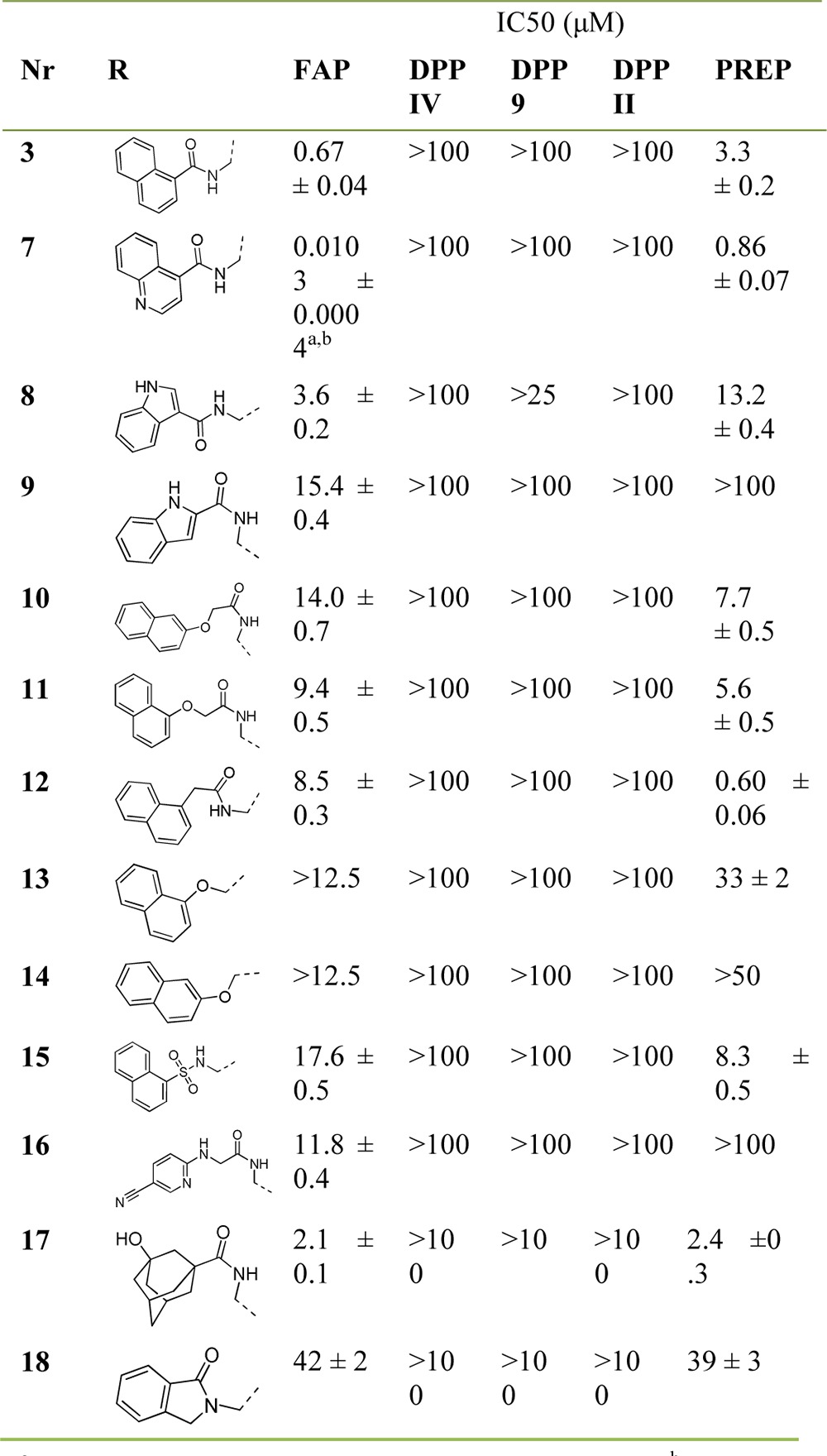
FAP-affinity (Ki) was determined to be 3.0 ± 0.4 nM.
The mean result of three separate measurements.
In terms of selectivity, all inhibitors in Table 2 display very limited to no affinity for the DPPs. As mentioned earlier, this can be rationalized by the absence of a basic P2 amine function known to be engaged in salt bridging with two acidic glutamate residues in the active center of the DPPs. The corresponding Glu203 and Glu204 in FAP have a different orientation to accommodate endopeptidase substrates, and thus also allow acylated P2-amines to enter FAP’s active center.15 As reported earlier, selectivity of N-acylated inhibitors with respect to the endopeptidase PREP is far less evident. Nonetheless, a comparison of initial ‘hit’ 3 (SI[FAP/PREP] = 5.3) and our new ‘lead’ 7 (SI[FAP/PREP] = 85) indicates that selectively optimizing for FAP affinity is possible.
Next, we turned our attention to the effect of substituting the N-(4-quinolinoyl) ring in 7 (Table 3). Overall, none of the evaluated substituents was able to improve FAP potency significantly, with substituents in the 2- and 3-position of the quinoline ring as in compounds 19–24, having a clearly negative effect on this parameter. Identifying the factors that cause this effect is not evident from these six analogues, but both steric (19, 22, and 23) and electronic (20, 21, and 24, with an increased electron density in the pyridine ring compared to 7) factors seem to be contributive. Remarkably, PREP-affinity is affected in a more intricate manner by these substitution types: the negative effect of 3-hydroxylation in 20 and 24 significantly contrasts with the influence of a 2-hydroxyl functionality in 21. Introducing a chlorine or bromine substituent at the 5-position of the quinoline ring such as in analogues 25 and 26 has limited effect on FAP potency. Most remarkably, the selectivity with respect to PREP of these inhibitors sharply increases (SI[FAP/PREP] > 1000), rendering them the most favorable affinity/selectivity profile of all FAP-inhibitors we had identified so far. Functionalizing the quinoline ring’s 6-position as in compounds 27–30 does not generally lead to marked differences in the affinity and selectivity profiles of the corresponding inhibitors compared to ‘lead’ 7. The most notable compound in this subseries of molecules is the 6-methoxyquinoline 30, which displays 10-fold less PREP affinity than its congeners. The 7-substituted chloroquinoline 31 and 7-bromoquinoline 32 again do not display relevantly changed FAP affinity when compared to 7, but they do have moderately increased selectivity toward PREP. Finally, 8-chloroquinoline 33 and 8-bromoquinoline 34 were found to possess significantly reduced FAP potency, possibly due to steric effects. These modifications, however, have little effect on the PREP inhibitory potential of the corresponding molecules.
Table 3. Effect of Substituents on the 4-Quinolinoyl Residue.
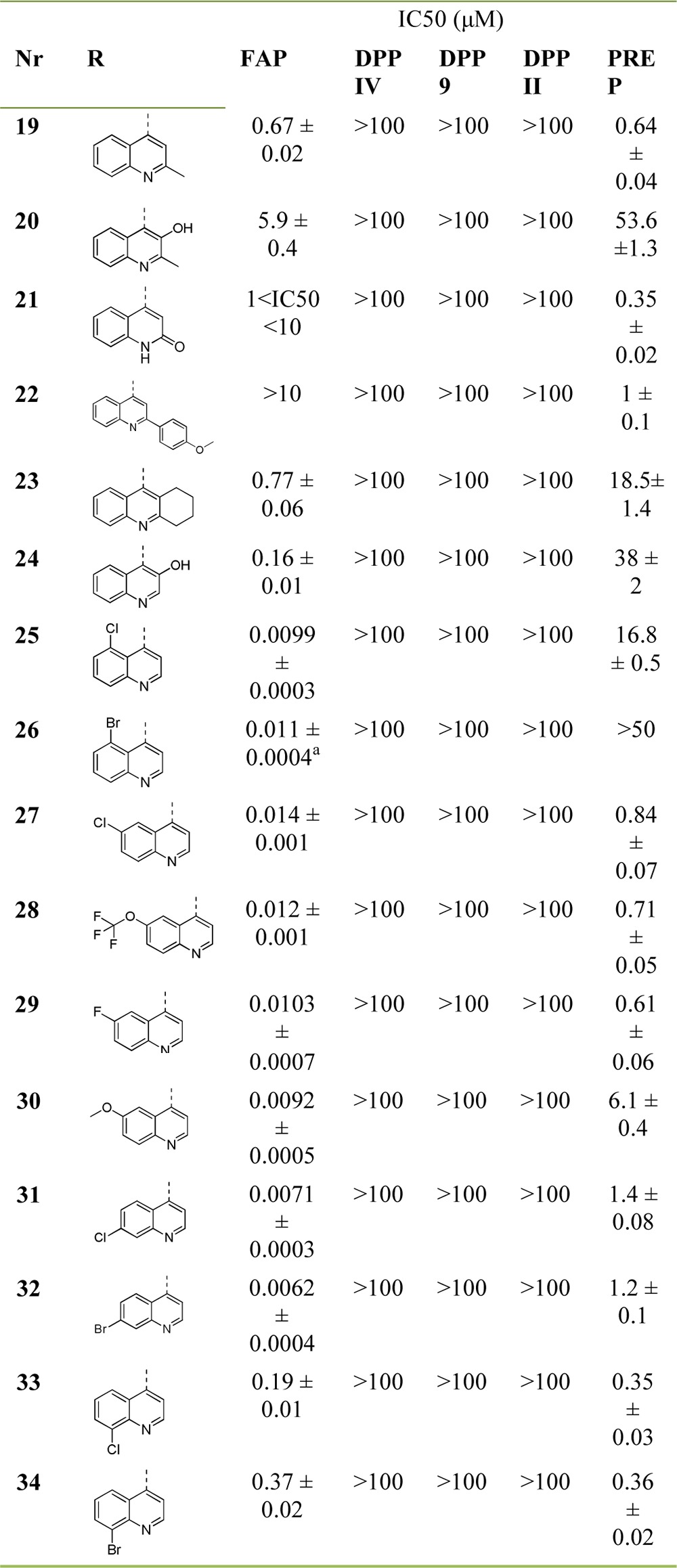
FAP-affinity (Ki) was determined to be 4.1 ± 0.6 nM.
We subsequently studied the effect of varying the position of the sp2 hybridized nitrogen atom of the quinoline ring (Table 4). We considered these investigations highly instrumental for understanding the striking difference in FAP affinity between ‘hit’ 3 and ‘lead’ 7. If inhibitor affinity would depend significantly on the ring nitrogen’s position, this could be indicative for a specific interaction with the enzyme, other than the cation−π complexation we originally hypothesized. On the basis of our docking study, the favorable contribution to binding of the latter would be less dependent on the nature of the (iso-)quinolinoyl isomer selected.
Table 4. Effect of Nitrogen Position in the (Iso-)Quinolinoyl Ring.
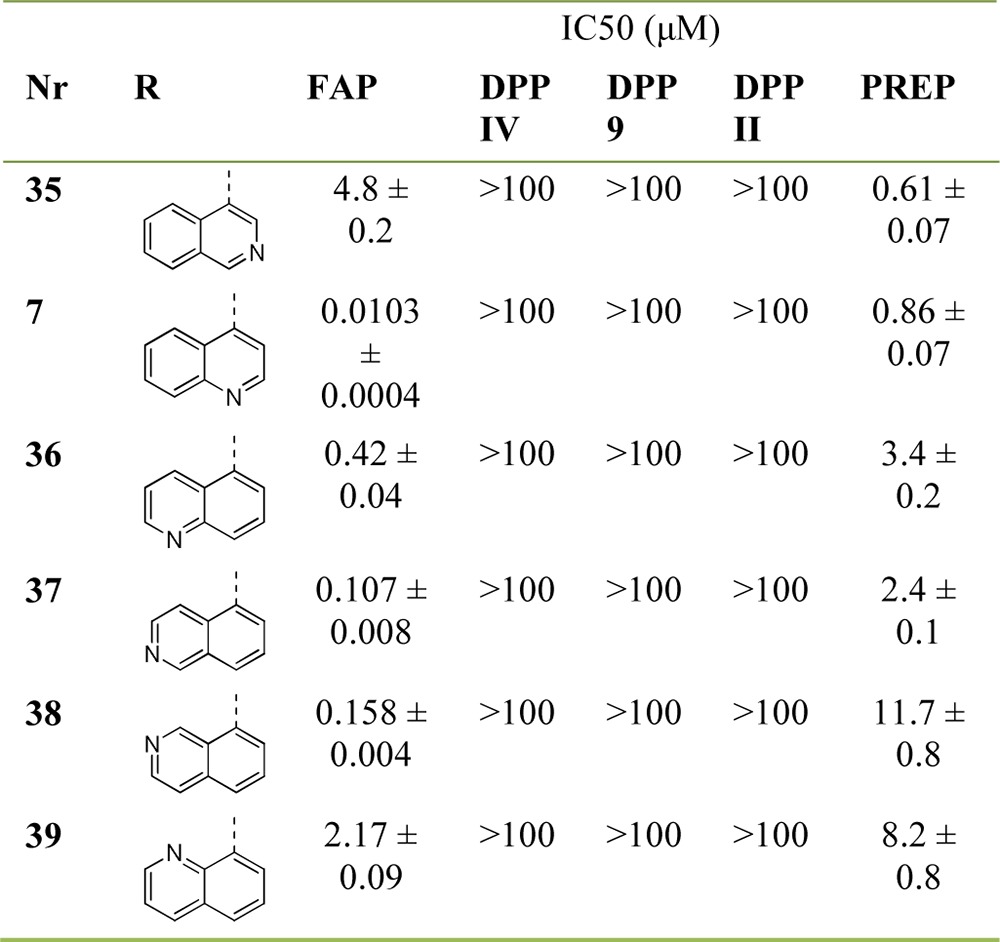
With FAP-affinities spanning almost 3 orders of magnitude, evaluation results of compounds 35–39 nonetheless reveal a pivotal importance of the nitrogen’s position. Of all the positional isomers synthesized, the 4-quinolinoyl ring of ‘lead’ 7 clearly displays the best results and takes in a singular position within this series. The 4-isoquinolinoyl and 8-quinolinoyl derivatives (35 and 39, respectively) are characterized by very low FAP-affinity, even when compared to ‘hit’ 3.
In conclusion, we have identified the N-(4-quinolinoyl)glycyl-(2-cyanopyrrolidine) scaffold as highly promising for discovery of FAP-inhibitors. It is the first scaffold type demonstrated to have potential for rendering compounds that combine low nanomolar FAP-affinity and a selectivity index >103 with respect to both the DPPs and PREP. Our SAR investigations so far have mainly focused on the P3 part of the scaffold but already allow to conclude that the N-(4-quinolinoyl) residue is a critical element in determining FAP-affinity. As such, it was found to hold a privileged position within the series of different quinolinoyl and isoquinolinoyl isomers evaluated. Furthermore, we have shown that introduction of substituents at the 5-position of the quinolinoyl ring system is a possible means of maximizing inhibitor selectivity with respect to PREP. So far, however, we have not been able to identify the eventual specific interactions between FAP and the 4-quinolinoyl ring that could underlie the observed affinities.
To this end, we are currently expanding our SAR-data set by investigating additional modification types in the P3, P2, and P1 region of these molecules. In addition, preclinical ADME and in vivo pharmacokinetic parameters of the optimal molecules will be determined. Inhibitor 7 has a kinetic solubility of >200 μM and a log D of 0.51 at pH 7.4. Furthermore, 7 was stable for >24 h both in PBS buffer at pH 7.4 and in rat plasma. Furthermore, 26, which together with inhibitor 25 shares low-nanomolar FAP-affinity and a FAP/PREP selectivity index >103, has a kinetic solubility of >200 μM and a log D of 0.7 at pH 7.4. This molecule was stable for 24 h in PBS and human plasma and 80% stable after 24 h in mouse plasma. It displayed mouse microsomal stability of 70% over 24 h.
Acknowledgments
We are indebted to Nicole Lamoen and Sophie Lyssens for excellent technical assistance.
Glossary
Abbreviations
- FAP
fibroblast activation protein
- PREP
prolyl oligopeptidase
- DPPII
dipeptidyl peptidase II
- DPPIV
dipeptidyl peptidase IV
- DPP9
dipeptidyl peptidase 9
- HATU
2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- SI
selectivity index
Supporting Information Available
Analytical data and experimental procedures for synthetic preparation, enzymatic evaluation, and physicochemical characterization of compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was financially supported by the Fund for Scientific Research Flanders/FWO-Vlaanderen (to I.D.M. and A-M.L.), the research (BOF)-fund of the University of Antwerp (to K.J., I.D.M., K.A., and P.V.d.V.), and the Hercules foundation.
The authors declare no competing financial interest.
Supplementary Material
References
- Aertgeerts K.; Levin I.; Shi L.; Snell G. P.; Jennings A.; Prasad G. S.; Zhang Y.; Kraus M. L.; Salakian S.; Sridhar V.; Wijnands R.; Tennant M. G. Structural and kinetic analysis of the substrate specificity of human fibroblast activation protein alpha. J. Biol. Chem. 2005, 280, 19441–19444. [DOI] [PubMed] [Google Scholar]
- Garin-Chesa P.; Old L. J.; Rettig W. J. Cell-surface protein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 7235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlan M. J.; Raj B. K.; Calvo B.; Garin-Chesa P.; Sanz-Moncasi M. P.; Healey J. H.; Old L. J.; Rettig W. J. Molecular cloning of fibroblast activation protein-alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 5657–5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos A. M.; Jung J.; Aziz N.; Kissil J. L.; Puré E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Invest. 2009, 119, 3613–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J. D.; Valianou M.; Canutescu A. A.; Jaffe E. K.; Lee H. O.; Wang H.; Lai J. H.; Bachovchin W. W.; Weiner L. M. Abrogation of fibroblast activation protein enzymatic activity attenuates tumor growth. Mol. Cancer Ther 2005, 4, 351–360. [DOI] [PubMed] [Google Scholar]
- Wang X. M.; Yao T.-W.; Nadvi N. A.; Osborne B.; McCaughan G. W.; Gorrell M. D. Fibroblast activation protein and chronic liver disease. Front. Biosci. 2008, 13, 3168–3180. [DOI] [PubMed] [Google Scholar]
- Acharya P. S.; Zukas A.; Chandan V.; Katzenstein A. L.; Pure E. Fibroblast activation protein: a serine protease expressed at the remodeling interface in idiopathic pulmonary fibrosis. Hum. Pathol. 2006, 37, 352–360. [DOI] [PubMed] [Google Scholar]
- Dienus K.; Bayat A.; Gilmore B. F.; Seifert O. Increased expression of fibroblast activation protein-alpha: implications for the development of a new treatment option. Arch. Dermatol. Res. 2010, 302, 725–731. [DOI] [PubMed] [Google Scholar]
- Milner J. M.; Kevorkian L.; Young D. A.; Jones D.; Wait R.; Donell S. T.; Barksby E.; Patterson A. M.; Middleton J.; Cravatt B. F.; Clark I. M.; Rowan A. D.; Cawston T. Fibroblast activation protein alpha is expressed by chondrocytes following a pro-inflammatory stimulus and is elevated in osteoarthritis. Arthritis Res. Ther. 2006, 8, R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner M.; Patel A.; Rowan A. D. Emerging role of serine proteinases in tissue turnover in arthritis. Arthritis Rheum. 2008, 58, 3644–3656. [DOI] [PubMed] [Google Scholar]
- Bauer S.; Jendro M. C.; Wadle A.; Kleber S.; Stenner F.; Dinser R.; Reich A.; Faccin E.; Gödde S.; Dinges H.; Müller-Ladner U.; Renner C. Fibroblast activation protein is expressed by rheumatoid myofibroblast-like synoviocytes. Arthritis Res. Ther. 2006, 8, R171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokopp C. E.; Schoenauer R.; Richards P.; Bauer S.; Lohmann C.; Emmert M. Y.; Weber B.; Winnik S.; Aikawa E.; Graves K.; Genoni M.; Vogt P.; Lüscher T. F.; Renner C.; Hoerstrup S. P.; Matter C. M. Fibroblast activation protein is induced by inflammation and degrades type I collagen in thin-cap fibroatheroma. Eur. Heart J. 2011, 32, 2713–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien P.; O’Connor B. F. Seprase: an overview of an important matrix serine protease. Biochim. Biophys. Acta 2008, 1784, 1130–1145. [DOI] [PubMed] [Google Scholar]
- Keane F. M.; Nadvi N. A.; Yao T.-W.; Gorrell M. D.; Neuropeptide Y. B-type natriuretic peptide, substance P and peptide YY are novel substrates of fibroblast activation protein-alpha. FEBS J. 2011, 278, 1326–1332. [DOI] [PubMed] [Google Scholar]
- Wolf B. B.; Quan C.; Tran T.; Wiesman C.; Sutherlin C. On the edge of vamidation-Cancer protease fibroblast activation protein. Mini-Rev. Med. Chem. 2008, 8, 719–727. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Ma L.; Wu M.; Wong M. S.; Li B.; Corral S.; Yu Z.; Nomanbhoy T.; Alemayehu S.; Fuller S. R.; Rosenblum J. S.; Rozenkrants N.; Minimo L. C.; Ripka W. C.; Szardenings A. K.; Kozarich J. W.; Shreder K. R. Synthesis and structure–activity relationship of N-alkyl-Gly-boroPro inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 4239–4242. [DOI] [PubMed] [Google Scholar]
- Eager R. M.; Cunningham C. C.; Senzer N. N.; Stephenson J.; Anthony S. P.; O’Day S. J.; Frenette G.; Pavlick A. C.; Jones B.; Uprichard M. Nemunaitis, Phase II assessment of talabostat and cisplatin in second-line stage IV melanoma J. BMC Cancer 2009, 9, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narra K.; Mullins S. R.; Lee H. O.; Stzremkowski-Brun R.; Magalong K.; Christansen V. J.; McKee P. A.; Egleston B.; Cohen S. J.; Weiner L. M.; Meropol N. J.; Cheng J. D. Phase II trial of single agent Val-boroPro (talabostat) inhibiting fibroblast activation protein in patients with metastatic colorectal cancer. Cancer Biol. Ther. 2007, 7, 1691–1699. [DOI] [PubMed] [Google Scholar]
- Ryabtsova O.; Jansen K.; Van Goethem S.; Joossens J.; Cheng J. D.; Lambeir A.-M.; De Meester I.; Augustyns K.; Van der Veken P. Acylated Gly-(2-cyano)pyrrolidines as inhibitors of fibroblast activation protein (FAP) and the issue of FAP/prolyl oligopeptidase (PREP)-selectivity. Bioorg. Med. Chem. Lett. 2012, 22, 3412–3417. [DOI] [PubMed] [Google Scholar]
- Tsai T. Y.; Yeh T. K.; Chen X.; Hsu T.; Jao Y. C.; Huang C. H.; Song J. S.; Huang Y. C.; Chien C. H.; Chiu Y. H.; Yen S. C.; Tang H. K.; Chao Y. S.; Jiaang W. K. Substituted 4-carboxymethylpyroglutamic acid diamides as potent and selective inhibitors of fibroblast activation protein. J. Med. Chem. 2010, 53, 6573–6583. [DOI] [PubMed] [Google Scholar]
- Villhauer E. B.; Brinkman J. A.; Naderi G. B.; Burkey B. F.; Dunning B. E.; Prasad K.; Mangold B. L.; Russell M. E.; Hughes T. E. 1-[[(3-Hydroxy-1-adamantyl)amino]acetyl]-2-cyano-(S)-pyrrolidine: a potent, selective, and orally bioavailable dipeptidyl peptidase IV inhibitor with antihyperglycemic properties. J. Med. Chem. 2003, 46, 2774–2789. [DOI] [PubMed] [Google Scholar]
- Augeri D. J.; Robl J. A.; Betebenner D. A.; Magnin D. R.; Khanna A.; Robertson J. G.; Wang A.; Simpkins L. M.; Taunk P.; Huang Q.; Han S. P.; Abboa-Offei B.; Cap M.; Xin L.; Tao L.; Tozzo E.; Welzel G. E.; Egan D. M.; Marcinkeviciene J.; Chang S. Y.; Biller S. A.; Kirby M. S.; Parker R. A.; Hamann L.G. S. Synthesis of novel potent dipeptidyl peptidase IV Inhibitors with enhanced chemical stability: interplay between the N-terminal amino acid alkyl side chain and the cyclopropyl group of α-aminoacyl-l-cis-4,5-methanoprolinenitrile-based inhibitors. J. Med. Chem. 2004, 47, 2587–2598. [DOI] [PubMed] [Google Scholar]
- Ashworth D. A.; Atrash B.; Baker G. A.; Baxter A. J.; Jenkins P. D.; Jones M. D.; Szelke M. 4-Cyanothiazolidides as very potent, stable inhibitors of dipeptidyl peptidase IV. Bioorg. Med. Chem. Lett. 1996, 6, 1163. [Google Scholar]
- Van der Veken P.; Haemers A.; Augustyns K. Prolyl peptidases related to dipeptidyl peptidase IV: Potential of specific inhibitors in drug discovery. Curr. Top. Med. Chem. 2007, 7, 621–635. [DOI] [PubMed] [Google Scholar]
- Thomas L.; Eckhardt M.; Langkopf E.; Mark M.; Tadayyon M.; Thomas L.; Nar H.; Pfrengle W.; Guth B.; Lotz R.; Sieger P.; Fuchs H.; Himmelsbach F. 8-(3-(R)-Aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2007, 50, 6450–6453. [DOI] [PubMed] [Google Scholar]
- Van Goethem S.; Mateeussen V.; Joossens J.; Lambeir A. M.; Chen X.; De Meester I.; Haemers A.; Augustyns K.; Van der Veken P. Structure–activity relationship studies on isoindoline inhibitors of dipeptidyl peptidases 8 and 8 (DPP8, DPP9): is DPP8-selectivity attainable?. J. Med. Chem. 2011, 54, 5737–5746. [DOI] [PubMed] [Google Scholar]
- Dubois V.; Van Ginneken C.; De Cock H.; Lambeir A. M.; Van der Veken P.; Augustyns K.; Chen X.; Scharpe S.; De Meester I. Enzyme activity and immunohistochemical localization of dipeptidyl peptidase 8 and 9 in male reproductive tissues. J. Histochem. Cytochem. 2009, 57, 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T.; Quan C.; Edosada C. Y.; Mayeda M.; Wiesmann C.; Sutherlin D.; Wolf B. B. Synthesis and structure–activity relationship of N-acyl-Gly-, N-acyl-Sar- and N-blocked-boroPro inhibitors of FAP, DPP4 and POP. Bioorg. Med. Chem. Lett. 2007, 17, 1438–1442. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.