


Abstract
The repair of toxic double-strand breaks (DSB) is critical for the maintenance of genome integrity. The major mechanisms that cope with DSB are: homologous recombination (HR) and classical or alternative nonhomologous end joining (C-NHEJ versus A-EJ). Because these pathways compete for the repair of DSB, the choice of the appropriate repair pathway is pivotal. Among the mechanisms that influence this choice, deoxyribonucleic acid (DNA) end resection plays a critical role by driving cells to HR, while accurate C-NHEJ is suppressed. Furthermore, end resection promotes error-prone A-EJ. Increasing evidence define Poly(ADP-ribose) polymerase 3 (PARP3, also known as ARTD3) as an important player in cellular response to DSB. In this work, we reveal a specific feature of PARP3 that together with Ku80 limits DNA end resection and thereby helps in making the choice between HR and NHEJ pathways. PARP3 interacts with and PARylates Ku70/Ku80. The depletion of PARP3 impairs the recruitment of YFP-Ku80 to laser-induced DNA damage sites and induces an imbalance between BRCA1 and 53BP1. Both events result in compromised accurate C-NHEJ and a concomitant increase in DNA end resection. Nevertheless, HR is significantly reduced upon PARP3 silencing while the enhanced end resection causes mutagenic deletions during A-EJ. As a result, the absence of PARP3 confers hypersensitivity to anti-tumoral drugs generating DSB.
INTRODUCTION
Double-strand breaks (DSB) produced by endogenous (normal cell metabolism, replication linked errors) or exogenous (chemotherapeutic drugs) genotoxic agents are considered as the most cytotoxic forms of deoxyribonucleic acid (DNA) damage. If unrepaired or inappropriately repaired, they will cause cell death or induce genomic instability and cancer (1). To counteract the effect of DSB, eukaryotic cells have evolved two highly efficient repair pathways: homologous recombination (HR) and nonhomologous end joining (NHEJ) (2). HR is initiated by the 5′–3′ resection of the DSB, a process mediated by the Mre11–Rad50–Nbs1 (MRN) complex in cooperation with CtIP that catalyses limited resection and the 5′–3′ exonuclease Exo1 that catalyses extensive resection (3). A host of other proteins has been shown to promote DNA end resection including among others BRCA1, WRN, SMARCAD (Fun30) and BLM (4–7). The 3′ single-stranded overhang produced is protected by phosphorylated replication protein A (RPA). RPA is then replaced by the recombinase RAD51 that with RAD54 will catalyze the search of homologous sequences and promote strand invasion of the template DNA. Because HR requires a homologous template, it is thought to operate in S and G2 phases of the cell cycle. NHEJ consists of two subpathways: the classical NHEJ pathway (C-NHEJ) and the alternative NHEJ process (A-EJ). C-NHEJ is initiated by the association of the Ku70–Ku80 heterodimer with DNA ends that serves as a scaffold for the assembly of the other NHEJ factors including Aprataxin polynucleotide kinase/phosphatase-like factor (APLF), DNA–PKcs, Artemis, Cernunnos/XLF and the XRCC4/DNA ligase IV complex (8,9). C-NHEJ is thought to process structural compatible ends and is active throughout the cell cycle (10–13). The alternative pathway (A-EJ) is initiated by an Mre11-mediated end-resection activity in a manner similar to HR and involves additional proteins such as PARP1, XRCC1, DNA ligase III and histone H1 (14–20). This process is highly mutagenic representing a major source of translocations. Recently, in addition to its important role in HR, BLM has been shown to prevent CtIP/Mre11-mediated long-range deletion during A-EJ (21). Similarly, BRCA1 has been proposed to stabilize Ku80 at broken ends thereby protecting from mutagenic A-EJ (22).
These different pathways compete for the repair of DSB. Thus, the choice of the appropriate repair pathway is pivotal and is the subject of intense investigations in the repair field. Several mechanisms have been shown to be determinant in directing repair toward HR or NHEJ including signaling pathways, chromatin modifications, the cell-cycle stage and the resection of DNA ends, the two latter are believed to commit cells to repair by HR (2,23). Evidence is building that the balance between BRCA1 and 53BP1 or between Ku80 and Mre11 influences DNA end resection and are therefore determinant of whether repair will occur through HR or NHEJ (24–29).
Recent studies have defined Poly(ADP-ribose) polymerase 3 (PARP3) as a novel player in cellular response to DSB (30). PARP3 has been described to interact with partners belonging to the NHEJ pathway including DNA–PKcs, DNA ligase IV, Ku70 and Ku80 and to accelerate XRCC4/DNA ligase IV-mediated ligation of chromosomal DSB in concert with APLF (31,32). Accordingly, PARP3 was found to be efficiently recruited to laser-induced DNA damage sites (33). It appears that the PAR-dependent interaction of APLF with PARP3 is important for the subsequent ATM-catalysed phosphorylation of APLF and its retention at damaged DNA (34). As a result, PARP3 depletion in human cells delays the repair of radio-induced DSB (33). Altogether, the current research is consistent with a cooperative role of PARP3 and APLF in the last steps of NHEJ. However, whether PARP3, that binds chromatin and DNA in vitro, also regulates early events of DSB repair still remains enigmatic (32,35).
In this study, we examined how the absence of PARP3 impacts on the repair of DSB by both HR or NHEJ. We used the radiomimetic drug bleomycin and the topoisomerase II inhibitor etoposide. We show that PARP3 expression is induced upon etoposide treatment. While we previously reported that its depletion in human cells or its disruption in mice had no impact on their survival when exposed to ionizing radiation (33), here we show that it significantly reduced their survival upon etoposide or bleomycin treatment. Notably, we found that the absence of PARP3 led to increased DNA end resection, but the rate of HR measured by using the HR-reporter substrate is compromised. Because Ku80 has been identified as a modulator of end resection, we focused our attention on this protein. We identified an association of PARP3 and Ku80 that is enhanced in response to DSB. PARP3 PARsylates Ku70–Ku80 and facilitates its recruitment to laser-induced DNA damage sites, at one hand to limit DNA end resection during HR, on the other to stimulate C-NHEJ. Consequently, PARP3 deficiency impaired the efficiency of accurate C-NHEJ while causing resection-mediated mutagenic deletions during A-EJ. In addition, we identify PARP3 as a novel regulator of the balance between BRCA1 and 53BP1 that also modulates DSB repair fate.
Together these findings unveil a key contribution of PARP3 together with Ku80 in the initial events of DSB repair driving the choice of the appropriate repair pathway. In addition, our results define PARP3 as an interesting therapeutic target to potentialize the cytotoxic effect of DSB-inducing chemotherapeutic drugs in cancer therapies.
MATERIALS AND METHODS
Reagents, antibodies and plasmids
Antibodies used include: rabbit anti-PARP-3, 4698 for immunofluorescence (1:1000), immunoprecipitation and western blot (1:10 000) (33); mouse anti-PARP1, EGT69 for western blot (1:10 000) (36); rabbit anti-phospho RPAS33 and anti-phospho RPAS4S8 for western blot (1:1000, Bethyl Laboratories) and immunofluorescence (1:2000); mouse anti-RPA32 for western blot (1:10 000, GeneTex); rabbit anti-RPA32 for immunofluorescence (1:2000, AbCam); rabbit anti-BRCA1 for immunofluorescence (1:500, Millipore); goat anti-Ku70 for western blot (1:500, santa Cruz); mouse anti-Ku80 for western blot (1:2000, AbCam); mouse anti-GFP for western blot (1:1000, Roche); rabbit anti-actin for western blot (1:500, Sigma); mouse anti-RAD54 for immunofluorescence (1:200, Abcam), rabbit anti-53BP1 for immunofluorescence (1:100, Novus Biologicals); rabbit anti-Mre11 for western blot (1: 5000, Novus Biologicals); mouse anti-HA.11 for western blot (1:1000, Covance); peroxidase-conjugated sheep anti-mouse (1:30 000, GE Healthcare); peroxidase-conjugated goat anti-rabbit (1:50 000, GE Healthcare) and peroxidase-conjugated rabbit anti-goat (1:20 000, Sigma) for ECL detection. Alexa Fluor-488 goat anti-rabbit IgG or Alexa Fluor-568 goat anti-rabbit IgG (1:1500, Molecular Probes), Alexa Fluor-488 goat anti-mouse IgG or Alexa Fluor-568 goat anti-mouse IgG (1:1500, Molecular Probes) in immunofluorescence studies. The PARP inhibitor Ku-0058948 has been described (37). Etoposide and bleomycin were purchased from Mylan Laboratories Inc. NU7441 was purchased from Selleckchem. YFP–Ku80 has been provided by D. Chen (University of Texas Southwestern Medical Center, Dallas, USA). The HA-I-SceI plasmid has been described (15).
Cell culture and siRNA-mediated depletion
Control (ctl) and PARP3-depleted (PARP3kd) MRC5 cells have been described previously (33). MDA-MB231 cells were grown in Roswell Park Memorial Institute medium (RPMI) medium supplemented with 10% fetal calf serum and 1% Gentamicin at 37°C in 5% CO2. MRC5 were grown in Dulbecco's modified Eagle's medium-1 g/l D-glucose supplemented with 20% fetal calf serum and 1% Gentamicin at 37°C in 5% CO2. The GCV6 cell line was derived from SV40-transformed GM639 human fibroblasts and contains the green fluorescent protein GFP-based and cluster of differentiation CD4-based substrates (15,38,39). GCV6 were cultured in Dulbecco's modified Eagle's medium-1 g/l D-glucose containing 10% fetal calf serum, 1% Gentamicin and supplemented with 3μg/ml blasticidin and 350 μg/ml neomycin to maintain the expression of CD4 and GFP respectively. The 3T3–GFP–Ku80 cells were grown in Dulbecco's modified Eagle's medium-1 g/l D-glucose containing 10% fetal calf serum, 1% Gentamicin and supplemented with 2 μg/ml puromycin to maintain the expression of GFP–Ku80. The U2OS DR–GFP reporter cells were used to measure HR as described (40,41).
Gene-specific siRNAs (ON_TARGET plus smart pool) for PARP3 (L-009297), BRCA1 (L-003461), CtIP (L-0011376), MRE11 (L-009271), Exo1 (L-013120) and negative control siRNA siCTL (D-001810) were obtained from Dharmacon. Cells were transfected with 50 nM siRNA using either JetPrime or Interferin (PolyPlus) according to the manufacturer's instructions and processed for the indicated experiments from 48 h to 72 h later. To confirm the down regulation of the targeted protein, the transfected cells were collected 48 h or 72 h after transfection for quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) or western blot analysis respectively.
Colony-forming assay
Exponentially growing cells were treated with etoposide (10 μM) or bleomycin (6 μg/ml) for 1 h, rinsed twice with phosphate buffered saline (PBS) and collected by trypsinization. SiRNA-depleted cells were exposed to the DNA damaging agents as above 72 h after siRNA transfection, rinsed twice with PBS and collected by trypsinization. Cells were seeded at 1500 cells in 100-mm culture dishes in triplicate. Ten days later, cells were fixed for 30 min in formaldehyde (3.7%), stained with crystal violet (0.1%) and colonies were scored. Statistical analysis were determined by ANalysis Of VAriance (ANOVA) tests as indicated by P values using StatView software.
SCE assay and chromosome analysis
Sister chromatid exchange (SCE) was carried out using a classical BrdU-labeling protocol. Briefly, cells were treated with Etoposide (2 μM) for 1 h, rinsed with PBS and incubated in culture medium supplemented with BrdU (15 μM) for two cell cycles. Metaphase spreads, obtained as previously described (42), were incubated with 50 μg/ml Hoechst 33258 for 10 min and exposed to 365 nm ultraviolet light (Fisher Bioblock Scientific) for 30 min. After washings, metaphases were stained with 4% Giemsa (10 min) and captured with an automated cytogenetic scanner workstation (MetaSystems). The mean percentages of SCE per chromosome ± sem were calculated from 30 metaphases per condition.
Indirect immunofluorescence microscopy
Immunofluorescence was performed essentially as described previously (43). Briefly, cells were grown on glass coverslips (1×105 cells/ six-well plates) for 48 h, either mock-treated or treated with 50 μM etoposide in triplicate and released in fresh medium for the indicated time points. Cells were washed twice with PBS 1X, fixed for 15 min at 25°C in 4% formaldehyde diluted in PBS 1X–0.1% Triton X-100–0.1% skimmed milk, and washed again three times for 10 min at 25°C with PBS 1X–0.1% Triton X-100–0.1% skimmed milk. Cells were then incubated overnight at 4°C with the appropriate primary antibodies as indicated. After three washes for 10 min at 25°C with PBS–0.1% Triton X-100–0.1% skimmed milk, cells were incubated with the appropriate Alexa-labeled secondary antibodies for 2 h at 25°C. DNA was counterstained with 4′,6-diamidino-2-phenylindol (DAPI; 25 ng/ml in PBS 1X) and slides were mounted in Mowiol. Images were captured using a Leica microscope (Leica Microsystems) equipped with an ORCA-ER chilled CCD camera (Hammamatsu) and the capture software Openlab (Improvision).
Cell extracts, immunoprecipitation and western blot
For whole cell extracts, exponentially growing cells were lysed by three cycles of freeze/thaw in lysis buffer (20 mM TrisHCl pH 7.5, 400 mM KCl, 5 mM dithithreitol (DTT), 0.5 mM Pefabloc, 20% glycérol, 0.1% NP40, and protease inhibitor complex (PIC, Roche)). For PARP3 analysis in whole cell extracts, cells were lysed in Radioimmunoprecipitation assay buffer (RIPA) buffer (50 mM Tris pH 8, 1% triton, 0.5% sodium deoxycholate, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 50 mM NaF, 20 mM sodium pyrophosphate pH 7.2, 1 mM sodium orthotovanadate, 1 mM Pefabloc, PIC) for 10 min at 4°C. After centrifugation at 14 000 g at 4°C for 15 min, cleared suspension was quantified by Bradford protein assay. Nuclear extracts were prepared according to Groisman et al. (44). Briefly, cells were suspended in hypotonic buffer (10 mM TrisHCl pH 7.3, 10 mM KCl, 1.5 mM MgCl2, 10 mM β-mercaptoethanol, 0.2 mM phenylmethanesulfonyl fluoride (PMSF)). After centrifugation at 2000 g at 4°C for 5 min, pellets were resuspended in extraction buffer (15 mM Tris-HCl pH 7.3, 0.4 M NaCl, 1 mM EDTA, 1 mM MgCl2, 10% glycerol, 10 mM β-mercaptoethanol and 10 mM PMSF). Samples were incubated on ice for 30 min and centrifuged at 16 000 g for 30 min at 4°C. The supernatant was used as the nuclear extract fraction. Equivalent amounts of proteins were analyzed by 10% SDS–PAGE and immunoblotting. For immunoprecipitation experiments, MDA-MB231 cells (7 × 106) were treated with 50 μM etoposide for 1 h and released in fresh medium. Two hours later, cells were collected and lysed by incubation in ice for 10 min in RIPA buffer (50 mM Tris pH 8, 1% triton, 0.5% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 50 mM NaF, 20 mM sodium pyrophosphate pH 7.2, 1 mM sodium orthotovanadate, 1 mM Pefabloc, PIC). After centrifugation at 14 000 × g at 4°C for 15 min, the cleared suspension was diluted in dilution buffer (150 mM NaCl, 20 mM Tris-HCl pH 7.5, 0.1% NP40, 0.5 mM Pefabloc). After a preclearing with Protein A Sepharose (GE Healthcare) for 30 min at 4°C, the cleared suspensions were incubated with either purified anti-PARP-3 (4698) or rabbit anti-mouse antibody as control overnight at 4°C, followed by 3 h incubation at 4°C with Protein A Sepharose. After centrifugation at 10 000 rpm at 4°C for 15 min, beads were washed once with diluted buffer (250 mM NaCl, 20 mM Tris-HCl pH 7.5, 0.1% NP40, 0.5 mM Pefabloc), and three times with diluted buffer containing 150 mM NaCl. Final pellets were resuspended in loading buffer and subjected to 10% SDS/PAGE and immunoblotting.
In vitro (ADP-ribosyl)ation assays
3T3–GFP–Ku80 cells (4 × 106) were lysed by three cycles of freeze/thaw in lysis buffer (20 mM TrisHCl pH 7.5, 400 mM NaCl, 5 mM DTT, 0.5 mM Pefabloc, 20% glycérol, 0.1% NP40, PIC, 250 nM Ku-0058948). After centrifugation at 10 000 rpm at 4°C for 20 min, cleared suspension were diluted five times in dilution buffer (20 mM TrisHCl pH 7.5, 150 mM NaCl, 0.5 mM Pefabloc, 0.1% NP40, PIC, and 250 nM Ku-0058948) and immunopurified using GFP-Trap®_A (Chromotek, Planneg-Martinsried, Germany) for 2–3 h at 4°C. Beads were washed twice with lysis buffer, twice in dilution buffer containing 400 mM NaCl without Ku-0058948 and once in Activity buffer (see below). For the in vitro assays, purified PARP-3 (1 μg) was incubated with equivalent amounts of immunopurified GFP-Ku80 or GFP for 25 min at 25°C in 100 μl of activity buffer (50 mM Tris-HCl pH 7.5, 1 mM dithiothreitol, 0.1 mg/ml bovine serum albumine) containing 50 μCi α-32PNAD+ (Perkin Elmer, 800 Ci/mmol) in the presence of 4 μg DNase I activated calf thymus DNA. When indicated Ku-0058948 was added 2 h before lysis and maintained throughout the experiment. The reaction was stopped by the addition of 500 μl of cold dilution buffer on ice, beads were washed four times with dilution buffer and resuspended in 20 μl Laemmli buffer. Reaction products were analyzed by gel electrophoresis on 4–20% gradient SDS–PAGE and autoradiography.
Cell-cycle analysis
Control (ctl) and PARP3kd MRC5 cells (1 × 106) were treated with 5 μM etoposide for 3h and released in fresh medium for the indicated time points. Cells were collected by trypsinization, washed with PGE (1X PBS, 1% glucose, 1 mM EDTA) and fixed in 70% ethanol in polyethylene glycol (PGE) for 24 h at 4°C. Cells were rehydrated in PGE for 3 h, treated for 30 min with 100 μg/ml RNase I, filtered and stained with propidium iodure at a final concentration of 50 μg/ml for 30 min at 37°C. Flow cytometry analysis was carried out using FACS calibur and Cell Quest Software (Becton Dickinson).
In vitro end-joining assay
Plasmid-based assay for in vitro end joining was performed as described (45). Nuclear extracts were prepared in triplicate from three spleens/sample recovered from eight-week old Parp3+/+ and Parp3−/− mice (33).
In vivo NHEJ and HR assay
NHEJ was performed essentially as described (15). Briefly, GCV6 cells (9 × 104) were first transfected with the indicated siRNAs (50 nM) for 48 h and then transfected with the HA-I-SceI expression vector (pCBASce) for an additional 48 h. Next, cells were washed with PBS 1X, harvested in PBS containing 50 mM EDTA for 5 min to detach the cells and collected in 500 μl PBS 1X. After centrifugation at 8000 × g at 4°C for 15 s, pellets were fixed with 2% paraformaldehyde (PFA) diluted in PBS for 20 min at room temperature and then PFA was diluted by addition of 500 μl PBS 1X. After centrifugation at 8000 × g at 4°C for 15 s, supernatants were removed and cells were stained for 45 min with 2.5 μl of the anti-CD4-PE antibody (MACS) diluted in PBS–BSA 1%. Cells were washed twice in PBS before FACS analysis. For sequence junction analysis, we performed polymerase chain reaction (PCR) on genomic DNA using the primers CMV-5, 5′-ATTATGCCCAGTACATGACCTTATG-3′ and CD4-int, 5′-GCTGCCCCAGAATCTTCCTCT-3′. PCR products were cloned into pCRTM4Blunt-TOPO® (Invitrogen), which allowed the isolation of individual clones, and sequenced (GATC Biotech).
HR was performed as previously described (40). Briefly, U2OS cells containing the HR reporter DR–GFP and the inducible ISceI-GR-mCherry were transfected with 50 nM of the indicated siRNA for 72 h. Next cells were treated with 100 mg/ml of triamcinolone acetonide (TA, Sigma) for 48 h to induce nuclear translocation of the ISceI-mCherry. Analyzing GFP-positive cells out of the mcherry-positive cells in FACS, analysis was done using the FACS calibur and Cell Quest Software (Becton Dickinson).
Laser microirradiation and time-lapse imaging
MDA-MB231 were grown on glass coverslips (3 × 103 cells/ six-well plates) for 72 h. Next, cells were first transfected with control or siPARP3 using JetPrime as detailed above for 48 h followed by a second transfection with 0.6 μg YFP–Ku80 using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Transfected cells were processed for microirradiation studies 17 h later. For local DNA damage induction, cells were sensitized by incubation in medium containing Hoechst 33342 (6.6 μg/ml) for 10 min. Microirradiation and time-lapse imaging was carried out using an iMIC microscope (Till Photonics) equipped with a Toptica Laser iBEAM 405 nm and an Olympus 60x (1.45 NA) objective. During acquisition, cells were maintained at 37°C in a 5% CO2 humidified atmosphere using an environmental control system (Life Imaging Services). YFP was excited at 491 nm with the monochromator Polychrome V (Till Photonics). For microirradiation, a preselected line within the nucleus was microirradiated for 10 ms with the power set to 20% of a 405 nm diode laser. The laser power measured at the end of the optic fiber was 23.5 mW. After microirradiation wide-field images were acquired at high speed (typically < 70 ms) for 4 s and then every 2 ms for additional 15 s on an EMCCD camera (Andor Technology) and analyzed using ImageJ software. For evaluation of the recruitment kinetics, mean fluorescence intensities of the irradiated region were corrected for loss of fluorescence over the time course and normalized to the pre-irradiation value. Data was fit by nonlinear regression and recruitment curves were generated using GraphPad Prism 5 (La Jolla, CA).
RESULTS
The depletion of PARP3 sensitizes cells to anti-tumoral drugs generating DSB
We and others have previously defined PARP3 as an important player in cellular response to DSB (30,32–34). To substantiate on these results, we decided to explore the abundance of PARP3 upon the induction of DSB using the topoisomerase II inhibitor etoposide in two independent cell lines (Figure 1A and Supplementary Figure S1A). Consistent with the role of PARP3 in DSB repair, etoposide treatment of MDA-MB231 and MRC5 cells caused a significant increase in the nuclear level of PARP3 visible upon treatment (Figure 1A and Supplementary Figure S1A, left panels, compare lanes 2 with lanes 1) and maintained up to 5 h after release of the cells in fresh medium (Figure 1A and Supplementary Figure S1A, left panels, lanes 3–6). In contrast, the expression of PARP1 remained rather unchanged. Although less efficiently, the etoposide-induced induction of PARP3 was also detected in the whole cell extracts of both cell lines (Figure 1A and Supplementary Figure S1A, right panels) precluding any protein relocalization.
Figure 1.

PARP3 responds to and promotes survival upon the induction of double-strand breaks. (A) Enhanced expression of PARP3 upon etoposide treatment. Upper left panel: MDA-MB231 cells were either mock-treated (lane 1) or exposed to etoposide (Eto, 50 μM) (lanes 2–6) for 3 h and then released in fresh medium for the indicated time points (0 h, indicates no release). Equivalent amounts of nuclear extracts were analyzed by western blotting using the indicated antibodies. Upper right panel, MDA-MB231 cells were either mock-treated (lane 1) or treated with etoposide (Eto, 50 μM, 3 h, lane 2) and equivalent amounts of total protein extracts were analyzed by western blotting as above. Lower panel: representative immunofluorescence pictures showing the nuclear staining of PARP3 (green) in treated (b and c, Eto, 50 μM, 3 h) or nontreated (a, NT) MDA-MB231 cells. In (c), the etoposide treatment and immunofluorescence were performed 48 h after transfection with siPARP3. Insets: nuclei are stained in blue with DAPI. (B) The depletion of PARP3 sensitizes cells to DSB-inducing agents. Clonogenic survival of bleomycin-treated (upper panel) or etoposide-treated (lower panel) control (ctl) and PARP3kd cells in the absence or in the presence of the PARP1 inhibitor Ku-0058948 (100 nM). Experiments were performed >3 times giving similar results. Mean values of triplicates ±SD are indicated. ***P< 0.001. The depletion of PARP3 in the PARP3kd cells was verified by qRT-PCR (Supplementary Figure S1Ba).
We previously reported that the depletion of PARP3 in human cells or its disruption in mice did not increase their long-term sensitivity to X-irradiation (33). Because X-irradiation produces only 1 DSB for 25 SSB (46), we postulated that an increase in the ratio of DSB versus single strand breaks (SSB) would be cytotoxic to PARP3-depleted cells. To validate this hypothesis, we compared the sensitivity of our control (ctl) and PARP3-depleted (PARP3kd) human cells to anti-tumoral drugs generating significant amounts of DSB by clonogenic survival assays (Figure 1B and Supplementary Figure S1Ba-b). We used the radiomimetic drug bleomycin generating an average of 1 DSB for 6 SSB and the topoisomerase II inhibitor generating an average of 1 DSB for 10 SSB (47–50). As expected, the depletion of PARP3 rendered cells hypersensitive to both drugs. A substantial cytotoxicity of both drugs was also seen in the murine Parp3−/− cells (Supplementary Figure S1Bc and data not shown). To evaluate the impact of PARP1, we also compared the sensitivity of the ctl and the PARP3kd cells to both drugs after chemical inhibition of PARP1 using the potent PARP inhibitor Ku-0058948 (100 nM, a concentration sufficient to inhibit PARP1 in vivo but not PARP3, data not shown, (33)). Ku-0058948 alone had no significant effect on the survival of either ctl or PARP3kd cells (Supplementary Figure S1Bd). While the additional inhibition of PARP1 further reduced the survival of the PARP3kd cells exposed to bleomycin (77% loss of survival after PARP inhibition) likely owing to the reduced PARP1-mediated repair of the SSBs produced, no significant additive effect is observed after exposure to etoposide likely generating weak amounts of SSB in the experimental conditions used.
Together, these findings emphasize a specific role of PARP3 in cellular response to DSB while PARP1 responds preferentially to SSB.
PARP3 promotes DSB repair by NHEJ
PARP3 was initially found to interact with partners belonging to the classical NHEJ pathway including DNA–PKcs, Ku70, Ku80 and APLF (31,32). To question whether PARP3 is needed for efficient NHEJ we first used a plasmid-based in vitro DNA end-joining assay (45) and measured the capacity of Parp3+/+ and Parp3−/− murine spleen nuclear extracts to perform end joining of Sma1- and EcoR1-linearized plasmids (Figure 2A). Whereas extracts from the Parp3+/+ cells support efficient end joining leading to the generation of multimeric forms of the plasmids visualized on agarose gels, this activity was reduced in Parp3−/− cells (by 35% for the end joining of blunt ends (Sma1) and by 27% for the end joining of protruding single-stranded ends (EcoR1)). These results and the previously published functional cooperation between PARP3 and APLF in DSB repair (32) thus suggest that PARP3 operates in NHEJ. We next aimed to investigate whether PARP3 is implicated in NHEJ repair at the chromosomal level in mammalian cells (Figure 2B). We used the previously described GCV6 cell line containing two types of intrachromosomal NHEJ-reporter substrates: the GFP-based substrate that permits analysis of the NHEJ on closely adjacent ends separated by only 34 bp and in which the GFP reporter is located 16 bp from the I-SceI restriction site and the pCOH–CD4 substrate to measure the NHEJ of two distant ends separated by 3.2 kb in which the CD4 reporter is located 211 bp from the I-SceI site (41) (Supplementary Figure S2A). Interestingly, the siRNA-mediated PARP3 silencing resulted in a significant reduction (2-fold reduction) in the end-joining efficiency of the GFP substrate compared to the cells transfected with the control siRNA, but did not impair the end-joining efficiency of the CD4 substrate (Figure 2B, left panel). A similar result was obtained using the GSC5 cell line containing the same constructs (41) (Supplementary Figure S2B). These results would suggest that PARP3 is important for the NHEJ of closely adjacent DSB but might not be involved in the end joining of distant DSB. Alternatively, these data can be explained by excessive DNA resection in PARP3-silenced cells resulting in the degradation of the GFP substrate due to frequent short or intermediate deletions (20–100 bp), while very long deletions (> 200 bp) that would degrade the CD4 reporter are rare events (38). Resection-mediated intermediate deletions at the resealed junctions have been characterized as the hallmark of error-prone A-EJ (38,51). To verify our hypothesis further and evaluate the impact of PARP3 on the accuracy of NHEJ, we analyzed the quality of the repair junctions by sequencing the end-joining patterns of the CD4 amplification products (Figure 2B, right panel and Supplementary Figure S2C). We found that 50% (17/34 events) of the clones transfected with control siRNA exhibited accurate end joining, whereas this frequency decreased to 26% (9/34 events) in the PARP3-silenced clones. Conversely, the number of deletions increased from 50% (17/34 events) in the control cells to 74% (25/34 events) in the PARP3-depleted cells. These results indicate a potential role of PARP3 in promoting accurate C-NHEJ, as previously described for Ku80 (38). Furthermore, the extent of deletions was significantly larger in the PARP3-silenced cells with a significant accumulation in deletions of an intermediate size (20–100 bp) compared to the control cells thereby suggesting that PARP3 probably helps to protect DNA ends from increased resection during A-EJ similarly to what will be observed during HR described later in this study (Figures 3 and 5).
Figure 2.
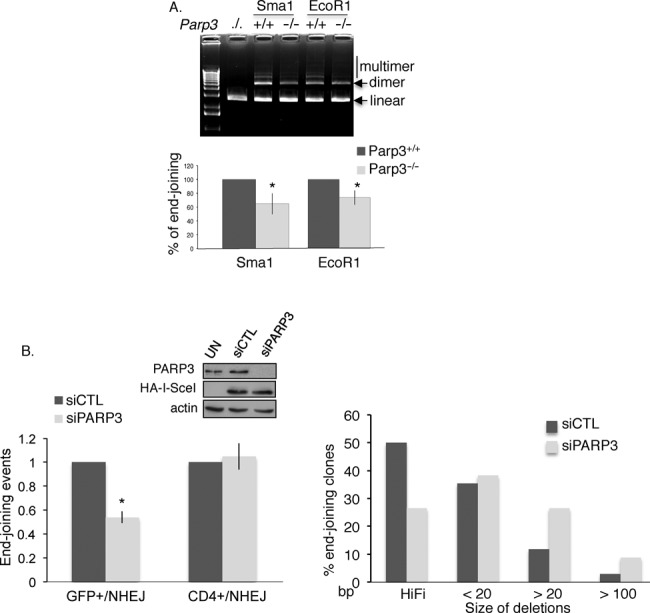
PARP3 promotes DSB repair by NHEJ. (A) Parp3−/− spleen cell extracts display reduced DNA end-joining activity. In vitro end joining of a Sma1 (blunt)- or EcoR1 (cohesive)-digested plasmid by nuclear cell extracts from Parp3+/+ and Parp3−/− splenocytes. Upper panel: agarose gel stained with ethidium bromide. Lower panel: the gels were scanned and signal intensities were measured using ImageJ. The value corresponds to the percentage of dimer and multimer products produced by Parp3−/− extracts relative to the Parp3+/+ extracts set to 100% and represents the mean of three independent experiments ±SD. *P< 0.05. (B) PARP3-depleted cells are defective in accurate NHEJ. Insets: PARP3 depletion and ISceI expression were verified by western blotting. Left panel: NHEJ efficiencies in the GCV6 cells treated with the indicated siRNAs. Values represent the means of three independent experiments (±SD). *P< 0.05. Right panel: analysis of the PCR patterns obtained by amplification of the CD4 fragment after NHEJ-mediated repair in control (siCTL) and PARP3-silenced (siPARP3) cells. Full-length PCR fragments represent high-fidelity end joining of the junction, whereas shorter PCR fragments indicate resection-mediated deletions, a hallmark of A-EJ. The silencing of PARP3 resulted in a decrease in high-fidelity end joining and an increase in the size of the deletions.
Figure 3.
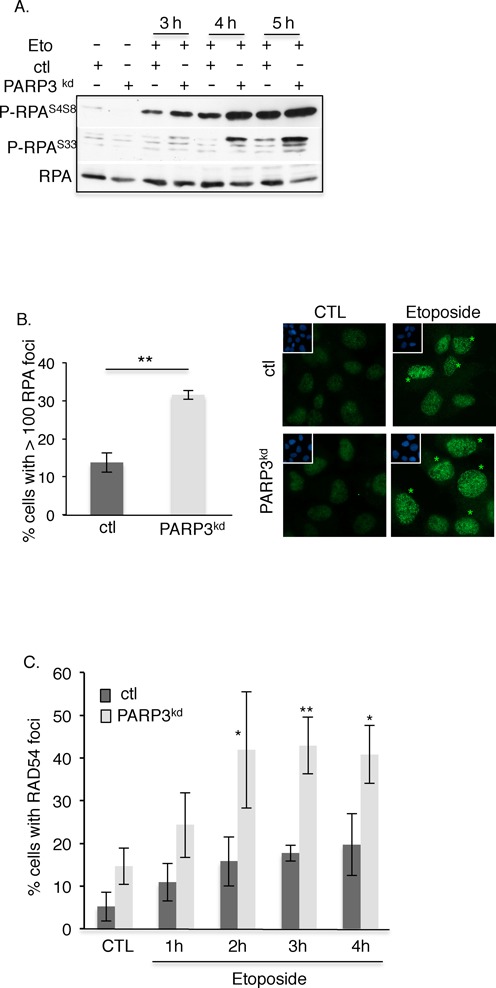
PARP3 limits extensive DSB end resection during HR. (A) Enhanced etoposide-induced phosphorylation of RPA32 in the absence of PARP3. Control (ctl) and PARP3-depleted cells (PARP3kd) were treated with etoposide (Eto, 50 μM), harvested at the indicated time points and processed for western blotting with antibodies against P-RPAS4S8, P-RPAS33 and RPA. (B) Increased etoposide-induced RPA foci formation in PARP3-depleted cells. Control (ctl) and PARP3-depleted cells (PARP3kd) were treated with etoposide (50 μM, 1 h), released in fresh medium for 5 h and processed for immunofluorescence using an anti-RPA antibody. Left panel: the histogram depicts the percentage of cells displaying > 100 RPA foci. An average of 500 cells per condition were scored in > 20 randomly selected fields. Mean values of three independent experiments ±SD are shown. **P< 0.01. Right panel: representative immunofluorescences of the quantification of the positive cells with RPA foci (green, asterisks) are shown. Insets: nuclei are stained in blue with DAPI. (C) Enhanced etoposide-induced loading of RAD54 in PARP3-depleted cells. Control (ctl) and PARP3-depleted cells (PARP3kd) were treated with etoposide (50 μM, 1 h) and processed for immunofluorescence at the indicated time points using an anti-RAD54 antibody. The histogram depicts the percentage of cells displaying RAD54 foci. An average of 500 cells per condition were scored in > 20 randomly selected fields. Mean values of three independent experiments ±SD are shown. *P < 0.05; **P< 0.01.
Figure 5.
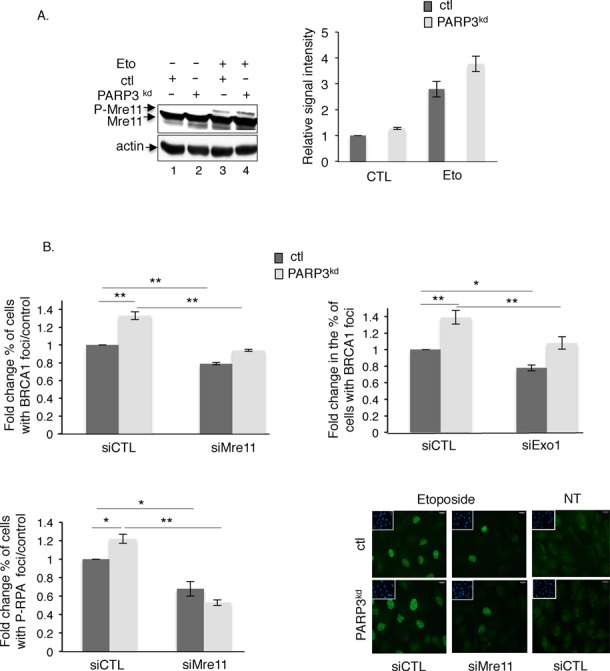
Extensive end resection in the PARP3-depleted cells is mediated by CtIP/Mre11 and Exo1 activities. (A) PARP3-depleted cells display increased etoposide-induced phosphorylation of Mre11. Control (ctl) and PARP3kd cells were mock-treated (lanes 1–2) or treated with etoposide (50 μM, 1 h, lanes 3–4) and harvested for Mre11 and actin immunoblotting (left panel). The DNA damage-induced phosphorylation of Mre11 induces a mobility shift that is enhanced in PARP3kd cells. Experiments were performed three times giving similar results (right panel). The relative signal intensities of the phosphorylated versus unphosphorylated Mre11 were measured in the three independent experiments using ImageJ. Mean values ±SD are indicated. (B) Extensive DNA end resection in the PARP3kd cells is partially rescued by the additional depletion of Mre11 or Exo1. Control (ctl) and PARP3-depleted cells (PARP3kd) were transfected with the indicated siRNA for 48 h, treated with etoposide (50 μM) for 1 h, released in fresh medium for 5 h and processed for BRCA1 (upper panel) and P-RPAS4S8 staining (lower panel). The histograms depict the fold change in the number of cells displaying >10 BRCA1 or >100 P-RPAS4S8 foci relative to the control. For each experiment, an average of 500 cells per condition were scored in > 20 randomly selected fields. Data are represented as the means of three independent experiments ±SD. *P < 0.05; **P< 0.01. Representative immunofluorescences of the quantification of the positive cells with P-RPAS4S8 foci (green) are shown. Insets: nuclei are stained in blue with DAPI. Note the similar pattern of cells with >100 P-RPAS4S8 foci in both cell lines.
In an attempt to further evaluate the relative contribution of PARP3 in both NHEJ subpathways, we also examined the sensitivity of the PARP3-depleted cells to etoposide in conditions where one or the other of the subpathway is compromised (Supplementary Figure S3). To inhibit the classical NHEJ pathway, we treated the ctl and PARP3kd cells with the DNA–PK inhibitor NU7441. To inhibit the alternative pathway, we examined the effect of the depletion of DNA ligase III, a key player of the A-EJ (16,52). While the depletion of PARP3 combined with the inhibition of DNA–PK further enhanced their sensitivity to etoposide, no additive effect was observed upon the combined depletion of PARP3 and DNA ligase III. Taken together, these findings suggest a role of PARP3 at the crossroad between classical and alternative end joining likely to limit mutagenic end resection thereby dictating the choice between both subpathways.
PARP3 prevents excessive DNA end resection during HR and controls the balance between BRCA1 and 53BP1
A drop in the efficiency of DSB repair by NHEJ is often associated with an increase in the rate of HR (41,53). Furthermore, end resection is critical for the generation of single-stranded DNA and the sequential loading of RAD51 during HR. To determine whether PARP3 also influences end resection during HR, we aimed to analyze how the depletion of PARP3 affects the etoposide-induced hyperphosphorylation of RPA, an event that was previously linked to efficient DNA end resection after genotoxic stress (54) (Figure 3A). We found strong increase in the etoposide-induced hyperphosphorylation of RPA on the residues S4S8 and S33 in the PARP3kd cells compared to the control cells indicating enhanced formation of DNA-damage-induced ssDNA regions. We also quantified the induction of both RPA and P-RPAS4S8 foci in response to etoposide in both cell lines and found a sizeable increase in the number of damaged cells containing RPA and P-RPA foci in the PARP3kd cells compared to the control cells (Figures 3B and 5B, lower panels). Interestingly, in both the ctl and PARP3kd cells, we obtained a very similar pattern with almost only a high number (>100) of small bright foci in the damaged cells. This observation is likely related to the accumulation of the cells in S phase at the time point studied (5 h release upon etoposide treatment) as detailed below (Figure 4C). Increased DNA end resection has been shown to induce the formation of a RAD51 nucleofilament on the 3′ overhang to initiate strand invasion on sister chromatids. However, we were unable to efficiently detect the etoposide-induced accumulation of RAD51 in our cell lines. Therefore, to confirm our observation further, we monitored the formation of RAD54 to nuclear foci, where it is thought to promote DNA topological changes aimed to facilitate homologous DNA pairing (55) (Figure 3C). In agreement with the RPA hyperphosphorylation, we observed enhanced RAD54 foci formation in PARP3kd cells upon exposure to etoposide compared to the control cells. Together, these data suggested that the absence of PARP3 induces elevated levels of DSB end resection during HR as observed above during A-EJ (Figure 2B).
Figure 4.
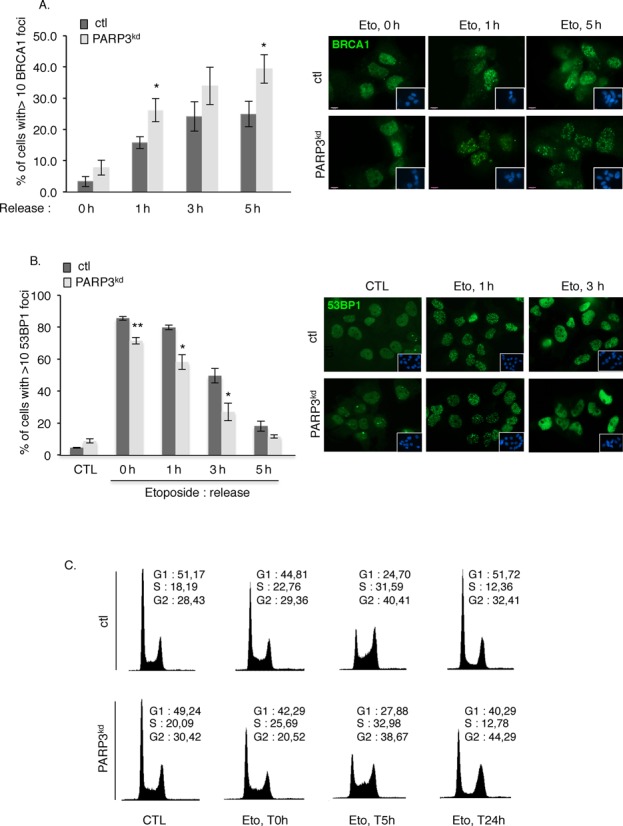
PARP3 helps to modulate the balance between BRCA1 and 53BP1. (A) Increased accumulation of BRCA1 at DSB sites in PARP3-depleted cells. Control (ctl) and PARP3-depleted cells (PARP3kd) were treated with etoposide (50 μM) for 1 h, released in fresh medium and processed for immunofluorescence at the indicated time points using an anti-BRCA1 antibody. The histogram depicts the percentage of cells displaying >10 BRCA1 foci. An average of 500 cells per condition were scored in > 20 randomly selected fields. Data are represented as the means of three independent experiments ±SD. *P < 0.05. Representative immunofluorescences of the quantification of the positive cells with BRCA1 foci (green) are shown. Insets: nuclei are stained in blue with DAPI. A similar pattern of BRCA1 foci was observed in both cell lines. (B) Decreased loading of 53BP1 in PARP3-depleted cells. Control (ctl) and PARP3-depleted cells (PARP3kd) were treated with etoposide (50 μM) for 1 h, released in fresh medium and processed for immunofluorescence at the indicated time points using an anti-53BP1 antibody. The histogram depicts the percentage of cells displaying >10 53BP1 foci. An average of 500 cells per condition were scored in > 20 randomly selected fields. Data are represented as the means of three independent experiments ±SD. *P < 0.05; **P< 0.01. Representative immunofluorescences of the quantification of the positive cells with 53BP1 foci (green) are shown. Insets: nuclei are stained in blue with DAPI. A similar pattern of 53BP1 foci was observed in both cell lines. (C) PARP3-depleted cells display a similar S-phase delay as control cells after etoposide treatment but a sustained G2/M arrest. FACS analysis of ctl and PARP3kd cells mock-treated (CTL) or treated with etoposide for 3 h (Eto, 5 μM) and released in fresh medium for the indicated time points (0 h (non released), 5 h, 24 h). y axis: cell numbers; x axis: relative DNA content based on propidium iodide staining.
The initiation of resection is a key determinant of repair pathway choice, which commits cells to HR and prevents repair by classical NHEJ. Genetic studies have established that this process is regulated by the balance between BRCA1 and 53BP1 at DSB chromatin (27,28). While the accumulation of BRCA1 at DSB promotes HR by displacing 53BP1 from DNA ends, the binding of 53BP1 triggers NHEJ presumably by inhibiting resection (56,57). Based on our above results, we wondered whether PARP3 modulates the competition between both proteins. To address this question, we analyzed the impact of PARP3 depletion on the assembly of etoposide-induced BRCA1 versus 53BP1 foci (Figure 4). Consistent with elevated end resection and impaired NHEJ, the formation of etoposide-induced BRCA1 foci was increased in PARP3kd cells compared to control cells (Figure 4A), while the assembly of 53BP1 foci was reduced upon etoposide in the PARP3-depleted cells compared to the control cells (Figure 4B). Thus, PARP3 helps to maintain the correct balance between BRCA1-dependent HR and 53BP1-mediated NHEJ.
It has recently been proposed that the antagonism between BRCA1 and 53BP1 is at least partly influenced by the cell cycle with the accumulation of 53BP1 at DSB restricted to G1 and the binding of BRCA1 prevailing from S phase onward (58). Furthermore, DSB resection must be appropriately restricted to S/G2, as HR requires an intact sister chromatid to promote repair. To determine a potential contribution of the cell cycle in the extensive end resection, we compared the cell-cycle distribution after etoposide-induced DNA damage in the control versus the PARP3kd cells (Figure 4C). We observed a comparable S-phase arrest 5 h post-treatment in both cell lines indicating that the extensive end resection observed in the PARP3kd cells cannot simply be explained by a higher accumulation in S phase of these cells. In contrast, we observed a more pronounced G2/M accumulation 24 h post-treatment in the PARP3kd versus the control cells that can be associated with a higher accumulation of unrepaired DNA strand breaks (33).
End resection is a two step process during HR initiated by Mre11 in cooperation with CtIP that carries limited end resection that is followed by extensive resection catalyzed by the 5′–3′ exonuclease Exo1 alone or in conjunction with the helicases WRN or BLM (59). Previous reports demonstrated that Mre11 goes through phosphorylation upon DNA damage, a post-translational modification required for its accumulation at repair foci (60,61). To verify that the increased end resection detected in the PARP3kd cells is caused by induced activation of Mre11, we followed the etoposide-induced phosphorylation of Mre11 visualized by a mobility shift of the protein. Consistent with the extensive DNA end resection, we detected a higher DNA damaged-induced phosphorylation of Mre11 in the PARP3kd cells compared to the ctl cells (Figure 5A, compare lane 4 with lane 3). No phosphorylation was detected in the untreated cells (lanes 1–2).
To substantiate on these results and confirm that the enhanced DNA end resection is mediated by either CtIP/Mre11 or Exo1, we tested whether the siRNA-mediated knockdown of either Mre11, CtIP or Exo1 would correct the excessive end resection detected in the PARP3kd cells (Figure 5B, upper panel and Supplementary Figure S4). We followed the formation of BRCA1 that accumulates at sites of DNA damage-induced ssDNA (7), 5 h upon recovery from etoposide treatment, a time point where we observed a significant difference between ctl and PARP3kd cells (Figure 4A, upper panel). As expected, the knockdown of either Mre11, CtIP or Exo1 significantly reduced the increased etoposide-induced accumulation of BRCA1 detected in the PARP3kd cells to the level of untreated control cells, thereby supporting the conclusion that PARP3-depleted cells display enhanced strand resection. The fact that the depletion of Mre11 only weakly (but significantly) reduces the loading of BRCA1 might be explained by a remaining resection activity (Supplementary Figure S4B) or various other pathways promoting BRCA1 recruitment (62–64). Therefore, to validate these observations further, we also measured the accumulation of P-RPAS4S8 that loads to the single-stranded tails downstream of BRCA1 (Figure 5B, lower panel). Similarly to BRCA1 foci formation, the knockdown of Mre11 markedly reduced the accumulation of P-RPAS4S8 induced by etoposide in both the ctl and PARP3kd cells.
Taken together, these data provide evidence for an involvement of PARP3 in the interplay between BRCA1 and 53BP1 and the regulation of ssDNA resection that is determinant of whether DSB repair ensures by either HR or NHEJ.
PARP3-depleted cells display reduced homology-directed repair of DSB
We next examined the consequence of the knockdown of PARP3 on the efficiency of HR-mediated DNA repair. To this end, we utilized the U2OS cell lines containing the stably integrated DR-GFP reporter and measured the HR efficiency induced by ISceI expression after the siRNA-mediated silencing of PARP3 (Figure 6A). Surprisingly, despite enhanced DNA end resection the reduction of PARP3 induced a significant drop in HR-mediated gene conversion compared to the one observed after depletion of the HR protein BRCA1 used as a positive control (9-fold decrease in BRCA1 silenced cells and 3.5-fold decrease in PARP3 silenced cells). HR between sister chromatids is the primary mechanism for SCE induced by genotoxic stress (65–70). Thus to validate this observation further, we tested whether the reduction of PARP3 would impair etoposide-induced SCE formation. We performed this experiment in siRNA-mediated PARP3-silenced MRC5 and MDA-MB231 cells compared to cells transfected with the control siRNA (siCTL) (Figure 6B and Supplementary Figure S5). In line with defective HR, we observed a reduced formation of SCE after exposure to etoposide in both PARP3-depleted versus control cells. Together, these data revealed impaired HR in PARP3-deficient cells despite enhanced end-resection activity.
Figure 6.

HR-mediated DSB repair is impaired by the depletion of PARP3. (A) HR efficiency is decreased after PARP3 depletion. The frequency of HR-mediated repair events was analyzed in U2OS+DR-GFP cells after transfection with the indicated siRNA and nuclear translocation of ISceI induced by exposure to Triamcinolone Acetonide (TA). The values correspond to the fold decrease of HR efficiency relative to the control (siCTL) and represent the means of three independent experiments ±SD. **P< 0.01; ***P< 0.001. (B) Etoposide-induced SCE formation is reduced in PARP3-depleted cells. SCE formation was analyzed in undamaged or etoposide-treated (+Eto) MDA-MB231 cells after transfection with the indicated siRNA. ***P< 0.001.
PARP3 interacts with Ku80 and facilitates its recruitment to DNA damaged sites
Previous studies using overeexpressed proteins have identified an interaction of PARP3 with Ku80 (31). Ku80 was reported to play a determinant role in dictating the choice between the repair pathways through the regulation of DNA end resection. After binding to DSB, Ku70/Ku80 must be actively displaced to allow DNA end resection and thus repair to occur. Its prolonged persistence at break sites was found to impair HR and accurate NHEJ while similarly to what we observed in our PARP3kd cells, its depletion was associated with higher level of resection partially dependent on Exo1 during HR and increased resection-mediated deletional end joining during A-EJ (26,38,53,71). Furthermore, BRCA1 was found to cooperate with Ku80 at DSB sites and Mre11 has been implicated in Ku removal (22). Based on these different observations together with the increased etoposide-induced accumulation of BRCA1 foci and the enhanced phosphorylation of Mre11 detected in the absence of PARP3 (Figure 4A and Figure 5A), we speculated that the relationship between PARP3 and Ku80 might be important to limit extensive end resection and promote efficient DSB repair. To address this question, we first examined the association of both partners upon DNA damage (Figure 7A). Untreated or etoposide-treated whole cell extracts were immunoprecipitated with an anti-PARP3 antibody or an irrelevant antibody and coimmunoprecipitation of Ku80 was assessed by western blotting. We detected enhanced association of PARP3 with Ku80 upon DSB induction (Figure 7A, compare lane 3 with lane 2). No unspecific immunoprecipitation of Ku80 with the control antibody was detected (lane 1).
Figure 7.
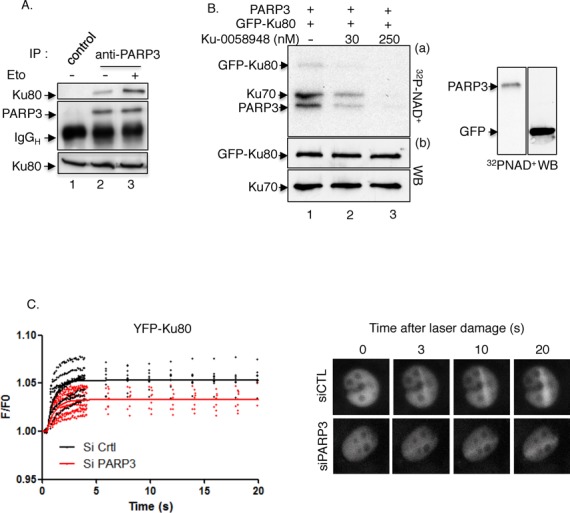
PARP3 interacts with and PARsylates Ku70–Ku80, and facilitates its recruitment to laser-induced DNA damage sites. (A) Enhanced association of PARP3 and Ku80 upon etoposide treatment. MDA-MB231 cells were mock-treated (lanes 1–2) or exposed to etoposide (50 μM) for 1 h and released in fresh medium for 2 h (lane 3). Cell extracts were immunoprecipitated with a control antibody (lane 1) or an anti-PARP3 antibody (lanes 2–3) and analyzed by western blotting using successively anti-Ku80 and anti-PARP3 antibodies. Input corresponds to 1/16 of the total amount of cell extracts used for immunoprecipitation. (B) PARP3 PARsylates the Ku70-Ku80 dimer. Immunopurified GFP–Ku80 was incubated with purified PARP3 and assayed for PARP activity in the absence (lane 1) or in the presence of Ku-0058948 (lanes 2–3). In similar experimental conditions, no PARsylation of GFP alone was detected. (C) Reduced recruitment of YFP-Ku80 at laser-induced DNA damage sites in PARP3-depleted cells. Left panel: MDA-MB231 cells were treated with the indicated siRNA for 48 h, transfected with YFP–Ku80 for 17 h and processed for laser microirradiation and live cell imaging as detailed in the Materials and Methods section. The difference in average fluorescence intensity in the damaged versus undamaged region is plotted against time. Right panel: a representative example of mock-depleted (siCTL) and PARP3-depleted (siPARP3) cells before and after microirradiation is shown.
To gain insights into the functional interaction between both partners, we evaluated the ability of PARP3 to PARsylate Ku80 (Figure 7B). Immunopurified GFP–Ku80 or GFP alone as a control was incubated together with purified PARP3 in the presence of α-32PNAD+ and DNase-I-treated calf thymus DNA. We found that PARP3 was able to weakly PARsylate GFP–Ku80 and significantly the coprecipitating Ku70 but not GFP alone (lane 1). The inhibition of PARP3 using the PARP inhibitor Ku-0058948 abolished the PARylation of Ku70 and GFP–Ku80 (lanes 2–3).
To explore the biological relevance of PARP3–Ku80 interaction, we analyzed how PARP3 depletion affects the accumulation of YFP–Ku80 to laser-induced DNA damage sites (Figure 7C). In agreement with the enhanced DSB end resection, we found that YFP–Ku80 recruitment was attenuated in PARP3-silenced cells compared to control cells indicating that PARP3 likely facilitates the recruitment of Ku80 to DSB to antagonize DNA end resection but facilitate Ku-mediated accurate classical NHEJ.
DISCUSSION
In earlier studies, we and others identified PARP3 as a newcomer in cellular response to DSB induced by ionizing radiations (32–34). Mechanistically, PARP3 was proposed to facilitate the phosphorylation and association of APLF with damaged DNA, which in turn accelerates the XRCC4/ligaseIV-mediated ligation during NHEJ (32,34). Whether PARP3 which interacts with the initial sensor of DSB, namely the Ku70/Ku80 heterodimer (31), also acts in early events of DSB repair driving the repair pathway choice remains unknown. Furthermore, the yield of irradiation-induced DSB is weak; it is estimated that 1 DSB is created for 25 SSB. As a result, despite delayed repair of radiation-induced DSB, long-term sensitivity was not affected by the absence of PARP3 (33). To clarify these issues, we analyzed the response of PARP3 to genotoxic compounds inducing significant levels of DSB and we questioned how PARP3 influences DSB-induced repair by HR versus NHEJ pathways.
We document that the nuclear abundance of PARP3 is greatly increased upon etoposide-induced DSB. Consistently, the long-term sensitivity of the PARP3-depleted cells to genotoxic agents increases with the DSB/SSB ratio. While X-irradiation had no impact (33), the long-term survival of PARP3-depleted cells was significantly compromised upon exposure to bleomycin and etoposide. Interestingly, we also observed an additive cytotoxic effect of PARP1 inhibition that seems more important upon exposure to bleomycin than to etoposide. We explain this difference by a higher accumulation of unrepaired SSB in bleomycin-treated cells. Together, these data are in strong support of a specific role of PARP3 in DSB repair while PARP1 responds to both SSB and DSB (72–75).
Our data also reveal a participation of PARP3 in the balance between competing DSB repair pathways. The resection of DSB to generate single-stranded DNA is a major restriction point in the choice between HR and C-NHEJ and is in competition with DNA end protection (2,23). In addition, end resection is a hallmark of mutagenic A-EJ (59). Several processes and proteins modulate these events. Among them, the association of Ku70–Ku80 to DNA ends favors end protection and promotes accurate C-NHEJ through the recruitment of the core components of the repair pathway (10,76). Several studies have shown that the absence of Ku80 not only impairs accurate NHEJ but also enhances end resection catalyzed by Exo1 and consequently HR-directed repair (53,71,77). Similarly, we find that PARP3 depleted-cells display deficient NHEJ, but enhanced DNA end resection that is marked by increased DNA-damage-induced accumulation of RPA and RAD54 foci together with accelerated RPA phosphorylation and increased activation of Mre11. Furthermore, extensive end resection caused increased deletional end joining during A-EJ as observed in Ku80-deficient cells (38). Earlier studies have identified an association of PARP3 with Ku80 (31). Here we show that this interaction is increased after the induction of DSB and that PARP3 PARsylates the Ku70–Ku80 heterodimer. Together these data provide evidence for a regulatory role of PARP3 in the repair pathway choice partly by facilitating the recruitment of Ku80 to DNA damage sites, thereby preventing extensive DNA end resection. The diminished recruitment of Ku80 to DNA-damaged sites that we observed upon PARP3 silencing might at one side increase and/or stabilize the binding of CtIP/Mre11 and Exo1 at DSB sites and consequently stimulate DSB resection and on the other side compromise efficient and accurate Ku-dependent NHEJ. Supporting these results and by analogy to PARP3 in vertebrates, genetic studies in Dictyostelium identified a third ADP-ribosyltransferase termed Adprt1a that promotes the recruitement and/or retention of Ku80 to DSB to facilitate NHEJ and prevent HR (78). Accordingly, disruption of Adprt1a decreased the ability of cells to perform end joining with a concomitant increase in HR. We have yet to determine how the poly(ADP-ribosyl)ation of Ku70–Ku80 regulates these events. It has also been reported that PARP3 promotes NHEJ via the recruitment of APLF at damaged sites, which in turn promotes the recruitment/retention of XLF and XRCC4–Lig4 for efficient DNA ligation during C-NHEJ (32). In this molecular complex, Ku80 plays a critical role in retaining and stabilizing APLF and consequently XRCC4–Lig4 into the Ku–DNA complexes (79). The participation of PARP3 in the recruitment of Ku80 and its poly(ADP-ribosyl)ation reported here define PARP3 as a key player in the assembly of Ku complexes during C-NHEJ. As proposed above, this would also help to prevent extensive resection during HR or A-EJ as it was suggested for Ku80 (53,80). However, the exact molecular mechanism involved remains to be clarified. One possibility is that the PARP3-mediated retention of Ku80 and thereby APLF and XRCC4/lig4 is a way to limit the access of resection enzymes to the break. In line with this hypothesis, Ku80 was shown to inhibit the recruitment of the resection enzyme Exo1 in human cells and to compete with the MRX/N resection factors (71,77,81).
Consistent with a regulatory role of PARP3 in the repair pathway choice, we also found that the absence of PARP3 affects the balance between BRCA1 and 53BP1 at DSB. Because histone modifications and the chromatin context are essential determinants of the competition between both proteins for DSB chromatin association (82,83), it is tempting to speculate on a possible involvement of PARP3 in modulating the chromatin state flanking DSB. In line with this idea, PARP3 has been shown to PARylate histones in vitro (32,84,85). It will be interesting in future studies to identify the histones PARylated by PARP3 at DNA lesions and determine how this influences the pattern of histone modifications and orchestrates the assembly of the repair factors at sites of damaged DNA. For example, it has previously been proposed that histone acetylation regulates the binding of Ku70/Ku80 at DSB sites (86,87). Similarly, it is possible that the PARP3-mediated modification of the chromatin environment at DSB is a way to modulate the binding and or retention of Ku70/Ku80 to damaged sites. Furthermore, PARP3 has been described to interact with the chromatin-associated Polycomb Group (PcG) components EZH2, Suz12 and YY1 (31). Beside their role in epigenetic gene silencing, PcG proteins have functional relevance in DNA damage repair and genome maintenance (88). They have been shown to confer protection against ionizing radiation and accumulate at DNA lesions in a poly(ADP-ribose)-dependent manner (89,90). However, the PARP member involved in this recruitment has not been clearly identified. PARP3 appears as an ideal candidate. EZH2 has been proposed to affect DSB repair through the regulation of BRCA1 (91). Given the increased accumulation of BRCA1 in the PARP3kd cells, it is possible that PARP3 cooperates with EZH2 to regulate the expression and/or binding of BRCA1 at sites of damaged DNA. An other possibility is that the interplay between PARP3 and PcG helps to coordinate DSB repair and transcription within the chromatin environment as it has been suggested for PARP1 (74,92).
That the sensitivity of the PARP3-depleted cells to genotoxic agents increases with the ratio of DSB/SSB also raises the possibility that its function in Ku80 recruitment and repair pathway choice is influenced by this ratio. At a biochemical level, this could be explained by a regulated cross talk between PARP3 in DSB resolution and PARP1 and/or PARP2 in SSB resolution. Forthcoming studies will help to clarify this hypothesis.
Surprisingly, despite extensive DNA end resection, HR as measured using the integrated GFP reporter substrate or by analyzing DNA-damage-induced SCE is severely compromised. Although the mechanism involved is unknown, these observations imply a blockage of the HR-mediated repair process at a step downstream of DNA end resection, during strand invasion as suggested by the higher accumulation of RAD54-containing cells, in the resolution or dissolution of the Holliday junctions or at the final ligation step. Furthermore, this apparent contradiction questions the interpretation whether extensive end resection might suppress HR. Alternatively, these results suggest a contribution of PARP3 to a later step of homology-dependent repair that is yet to be identified.
In summary, the work presented here provides good evidence that PARP3 limits end resection and thereby helps making the decision between HR and end-joining pathways (HR versus C-NHEJ and/or C-NHEJ versus A-EJ) partly in cooperation with the Ku70–Ku80 heterodimer, although the structure of the chromatin around the lesion may also be important. Furthermore, our findings may inspire the development of PARP3-specific inhibitors in therapeutic strategies aimed to potentialize the cytotoxic action of clastogenic drugs generating high amounts of DSB. Clearly, recent and ongoing structural and screening studies aimed to identify PARP3-specific inhibitors will be valuable in exploring such strategies (84,93).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online, including [94].
ACKNOWLEDGMENTS
The authors are grateful to M. Goldberg, Hebrew University, Israel for providing the U2OS DR-GFP-mCherry-I-SceI-GR reporter cells and David J. Chen, The University of Texas Southwestern Medical Center, Dallas, USA, for the YFP-Ku80 plasmid. We appreciate helpful discussions with E. Soutoglou, Department of Cancer Biology, IGBMC, Illkirch, France.
FUNDING
Agence Nationale de la Recherche; Association pour la Recherche contre le Cancer; Electricité de France; Ligue Nationale Contre le Cancer (Equipe Labellisée); Centre National de la Recherche Scientifique; Université de Strasbourg; National Institutes of Health [R01 GM073894 to R.S.]. This work has beenpublished within the LABEX ANR-10-LABX-0034_Medalis and received a financial support from French government managed by “Agence National de la Recherche” under “Programme d'investissement d'avenir”.
REFERENCES
- 1.Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapman J.R., Taylor M.R., Boulton S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 3.Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat. Struct. Mol. Biol. 2010;17:11–16. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costelloe T., Louge R., Tomimatsu N., Mukherjee B., Martini E., Khadaroo B., Dubois K., Wiegant W.W., Thierry A., Burma S., et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature. 2012;489:581–584. doi: 10.1038/nature11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gravel S., Chapman J.R., Magill C., Jackson S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nimonkar A.V., Ozsoy A.Z., Genschel J., Modrich P., Kowalczykowski S.C. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. U. S. A. 2008;105:16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlegel B.P., Jodelka F.M., Nunez R. BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res. 2006;66:5181–5189. doi: 10.1158/0008-5472.CAN-05-3209. [DOI] [PubMed] [Google Scholar]
- 8.Gottlieb T.M., Jackson S.P. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 9.Mahaney B.L., Meek K., Lees-Miller S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieber M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lieber M.R., Wilson T.E. SnapShot: Nonhomologous DNA end joining (NHEJ) Cell. 2010;142:496–496. doi: 10.1016/j.cell.2010.07.035. e491. [DOI] [PubMed] [Google Scholar]
- 12.Mladenov E., Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat. Res. 2011;711:61–72. doi: 10.1016/j.mrfmmm.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 13.Guirouilh-Barbat J., Huck S., Lopez B.S. S-phase progression stimulates both the mutagenic KU-independent pathway and mutagenic processing of KU-dependent intermediates, for nonhomologous end joining. Oncogene. 2008;27:1726–1736. doi: 10.1038/sj.onc.1210807. [DOI] [PubMed] [Google Scholar]
- 14.Rass E., Grabarz A., Bertrand P., Lopez B.S. [Double strand break repair, one mechanism can hide another: alternative non-homologous end joining] Cancer Radiother. 2012;16:1–10. doi: 10.1016/j.canrad.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Rass E., Grabarz A., Plo I., Gautier J., Bertrand P., Lopez B.S. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009;16:819–824. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 16.Audebert M., Salles B., Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 17.Wang M., Wu W., Rosidi B., Zhang L., Wang H., Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosidi B., Wang M., Wu W., Sharma A., Wang H., Iliakis G. Histone H1 functions as a stimulatory factor in backup pathways of NHEJ. Nucleic Acids Res. 2008;36:1610–1623. doi: 10.1093/nar/gkn013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paul K., Wang M., Mladenov E., Bencsik-Theilen A., Bednar T., Wu W., Arakawa H., Iliakis G. DNA ligases I and III cooperate in alternative non-homologous end-joining in vertebrates. PLoS One. 2013;8:e59505. doi: 10.1371/journal.pone.0059505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mansour W.Y., Rhein T., Dahm-Daphi J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010;38:6065–6077. doi: 10.1093/nar/gkq387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grabarz A., Guirouilh-Barbat J., Barascu A., Pennarun G., Genet D., Rass E., Grmann S., Bertrand P., Hickson I.D., Lopez B. A role for BLM in double strand break repair pathway choice : prevention of CtIP/Mre11-mediated alternative non-homologous end-joining. Cell Rep. 2013;5:21–28. doi: 10.1016/j.celrep.2013.08.034. [DOI] [PubMed] [Google Scholar]
- 22.Jiang G., Plo I., Wang T., Rahman M., Cho J.H., Yang E., Lopez B.S., Xia F. BRCA1-Ku80 protein interaction enhances end-joining fidelity of chromosomal double-strand breaks in the G1 phase of the cell cycle. J. Biol. Chem. 2013;288:8966–8976. doi: 10.1074/jbc.M112.412650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Symington L.S., Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 24.Barlow J.H., Lisby M., Rothstein R. Differential regulation of the cellular response to DNA double-strand breaks in G1. Mol. Cell. 2008;30:73–85. doi: 10.1016/j.molcel.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clerici M., Mantiero D., Guerini I., Lucchini G., Longhese M.P. The Yku70-Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep. 2008;9:810–818. doi: 10.1038/embor.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bunting S.F., Callen E., Kozak M.L., Kim J.M., Wong N., Lopez-Contreras A.J., Ludwig T., Baer R., Faryabi R.B., Malhowski A., et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell. 2012;46:125–135. doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunting S.F., Callen E., Wong N., Chen H.T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L., et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouwman P., Aly A., Escandell J.M., Pieterse M., Bartkova J., van der Gulden H., Hiddingh S., Thanasoula M., Kulkarni A., Yang Q., et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao L., Xu X., Bunting S.F., Liu J., Wang R.H., Cao L.L., Wu J.J., Peng T.N., Chen J., Nussenzweig A., et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell. 2009;35:534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boehler C., Dantzer F. PARP-3, a DNA-dependent PARP with emerging roles in double-strand break repair and mitotic progression. Cell Cycle. 2011;10:1023–1024. doi: 10.4161/cc.10.7.15169. [DOI] [PubMed] [Google Scholar]
- 31.Rouleau M., McDonald D., Gagne P., Ouellet M.E., Droit A., Hunter J.M., Dutertre S., Prigent C., Hendzel M.J., Poirier G.G. PARP-3 associates with polycomb group bodies and with components of the DNA damage repair machinery. J. Cell. Biochem. 2007;100:385–401. doi: 10.1002/jcb.21051. [DOI] [PubMed] [Google Scholar]
- 32.Rulten S.L., Fisher A.E., Robert I., Zuma M.C., Rouleau M., Ju L., Poirier G., Reina-San-Martin B., Caldecott K.W. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell. 2011;41:33–45. doi: 10.1016/j.molcel.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 33.Boehler C., Gauthier L.R., Mortusewicz O., Biard D.S., Saliou J.M., Bresson A., Sanglier-Cianferani S., Smith S., Schreiber V., Boussin F., et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. U. S. A. 2011;108:2783–2788. doi: 10.1073/pnas.1016574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fenton A.L., Shirodkar P., Macrae C.J., Meng L., Koch C.A. The PARP3- and ATM-dependent phosphorylation of APLF facilitates DNA double-strand break repair. Nucleic Acids Res. 2013;41:4080–4092. doi: 10.1093/nar/gkt134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Augustin A., Spenlehauer C., Dumond H., Menissier-De Murcia J., Piel M., Schmit A.C., Apiou F., Vonesch J.L., Kock M., Bornens M., et al. PARP-3 localizes preferentially to the daughter centriole and interferes with the G1/S cell cycle progression. J. Cell. Sci. 2003;116:1551–1562. doi: 10.1242/jcs.00341. [DOI] [PubMed] [Google Scholar]
- 36.Quenet D., Gasser V., Fouillen L., Cammas F., Sanglier-Cianferani S., Losson R., Dantzer F. The histone subcode: poly(ADP-ribose) polymerase-1 (Parp-1) and Parp-2 control cell differentiation by regulating the transcriptional intermediary factor TIF1beta and the heterochromatin protein HP1alpha. Faseb J. 2008;22:3853–3865. doi: 10.1096/fj.08-113464. [DOI] [PubMed] [Google Scholar]
- 37.Farmer H., McCabe N., Lord C.J., Tutt A.N., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C., et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 38.Guirouilh-Barbat J., Huck S., Bertrand P., Pirzio L., Desmaze C., Sabatier L., Lopez B.S. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol. Cell. 2004;14:611–623. doi: 10.1016/j.molcel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 39.Xie A., Hartlerode A., Stucki M., Odate S., Puget N., Kwok A., Nagaraju G., Yan C., Alt F.W., Chen J., et al. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol. Cell. 2007;28:1045–1057. doi: 10.1016/j.molcel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shahar O.D., Raghu Ram E.V., Shimshoni E., Hareli S., Meshorer E., Goldberg M. Live imaging of induced and controlled DNA double-strand break formation reveals extremely lowrepair of homologous recombination in human cells. Oncogene. 2012;31:3495–3504. doi: 10.1038/onc.2011.516. [DOI] [PubMed] [Google Scholar]
- 41.Lemaitre C., Fischer B., Kalousi A., Hoffbeck A.S., Guirouilh-Barbat J., Shahar O.D., Genet D., Goldberg M., Betrand P., Lopez B., et al. The nucleoporin 153, a novel factor in double-strand break repair and DNA damage response. Oncogene. 2012;31:4803–4809. doi: 10.1038/onc.2011.638. [DOI] [PubMed] [Google Scholar]
- 42.Gauthier L.R., Granotier C., Hoffschir F., Etienne O., Ayouaz A., Desmaze C., Mailliet P., Biard D.S., Boussin F.D. Rad51 and DNA-PKcs are involved in the generation of specific telomere aberrations induced by the quadruplex ligand 360A that impair mitotic cell progression and lead to cell death. Cell. Mol. Life Sci. 2012;69:629–640. doi: 10.1007/s00018-011-0767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dantzer F., Giraud-Panis M.J., Jaco I., Ame J.C., Schultz I., Blasco M., Koering C.E., Gilson E., Menissier-de Murcia J., de Murcia G., et al. Functional interaction between poly(ADP-Ribose) polymerase 2 (PARP-2) and TRF2: PARP activity negatively regulates TRF2. Mol. Cell. Biol. 2004;24:1595–1607. doi: 10.1128/MCB.24.4.1595-1607.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Groisman R., Polanowska J., Kuraoka I., Sawada J., Saijo M., Drapkin R., Kisselev A.F., Tanaka K., Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- 45.Iliakis G., Rosidi B., Wang M., Wang H. Plasmid-based assays for DNA end-joining in vitro. Methods Mol. Biol. 2006;314:123–131. doi: 10.1385/1-59259-973-7:123. [DOI] [PubMed] [Google Scholar]
- 46.Friedberg E., Walker G., Siede R., Wood R., Schultz R., Ellenberger T. DNA Repair and Mutagenesis. Washington, DC, USA: ASM Press; 2006. p. p. 1118. [Google Scholar]
- 47.Povirk L.F., Han Y.H., Steighner R.J. Structure of bleomycin-induced DNA double-strand breaks: predominance of blunt ends and single-base 5′ extensions. Biochemistry. 1989;28:5808–5814. doi: 10.1021/bi00440a016. [DOI] [PubMed] [Google Scholar]
- 48.Burden D.A., Osheroff N. Mechanism of action of eukaryotic topoisomerase II and drugs targeted to the enzyme. Biochim. Biophys. Acta. 1998;1400:139–154. doi: 10.1016/s0167-4781(98)00132-8. [DOI] [PubMed] [Google Scholar]
- 49.Long B.H., Musial S.T., Brattain M.G. DNA breakage in human lung carcinoma cells and nuclei that are naturally sensitive or resistant to etoposide and teniposide. Cancer Res. 1986;46:3809–3816. [PubMed] [Google Scholar]
- 50.Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guirouilh-Barbat J., Rass E., Plo I., Bertrand P., Lopez B.S. Defects in XRCC4 and KU80 differentially affect the joining of distal nonhomologous ends. Proc. Natl. Acad. Sci. U. S. A. 2007;104:20902–20907. doi: 10.1073/pnas.0708541104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simsek D., Brunet E., Wong S.Y., Katyal S., Gao Y., McKinnon P.J., Lou J., Zhang L., Li J., Rebar E.J., et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011;7:e1002080. doi: 10.1371/journal.pgen.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pierce A.J., Hu P., Han M., Ellis N., Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–3242. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mazin A.V., Mazina O.M., Bugreev D.V., Rossi M.J. Rad54, the motor of homologous recombination. DNA Repair (Amst) 2010;9:286–302. doi: 10.1016/j.dnarep.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bothmer A., Robbiani D.F., Feldhahn N., Gazumyan A., Nussenzweig A., Nussenzweig M.C. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 2010;207:855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chapman J.R., Sossick A.J., Boulton S.J., Jackson S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012;125:3529–3534. doi: 10.1242/jcs.105353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Escribano-Diaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J.T., Tkac J., Cook M.A., Rosebrock A.P., Munro M., Canny M.D., et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 59.Grabarz A., Barascu A., Guirouilh-Barbat J., Lopez B.S. Initiation of DNA double strand break repair: signaling and single-stranded resection dictate the choice between homologous recombination, non-homologous end-joining and alternative end-joining. Am. J. Cancer Res. 2012;2:249–268. [PMC free article] [PubMed] [Google Scholar]
- 60.Dong Z., Zhong Q., Chen P.L. The Nijmegen breakage syndrome protein is essential for Mre11 phosphorylation upon DNA damage. J. Biol. Chem. 1999;274:19513–19516. doi: 10.1074/jbc.274.28.19513. [DOI] [PubMed] [Google Scholar]
- 61.Yuan S.S., Chang H.L., Hou M.F., Chan T.F., Kao Y.H., Wu Y.C., Su J.H. Neocarzinostatin induces Mre11 phosphorylation and focus formation through an ATM- and NBS1-dependent mechanism. Toxicology. 2002;177:123–130. doi: 10.1016/s0300-483x(02)00220-2. [DOI] [PubMed] [Google Scholar]
- 62.Watanabe S., Watanabe K., Akimov V., Bartkova J., Blagoev B., Lukas J., Bartek J. JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1-mediated chromatin response to DNA breaks. Nat. Struct. Mol. Biol. 2013;20:1425–1433. doi: 10.1038/nsmb.2702. [DOI] [PubMed] [Google Scholar]
- 63.Yu H., Pak H., Hammond-Martel I., Ghram M., Rodrigue A., Daou S., Barbour H., Corbeil L., Hebert J., Drobetsky E., et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. U. S. A. 2013;111:285–290. doi: 10.1073/pnas.1309085110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee Y.H., Kuo C.Y., Stark J.M., Shih H.M., Ann D.K. HP1 promotes tumor suppressor BRCA1 functions during the DNA damage response. Nucleic Acids Res. 2013;41:5784–5798. doi: 10.1093/nar/gkt231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coleman K.A., Greenberg R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011;286:13669–13680. doi: 10.1074/jbc.M110.213728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chatterjee S., Trivedi D., Petzold S.J., Berger N.A. Mechanism of epipodophyllotoxin-induced cell death in poly(adenosine diphosphate-ribose) synthesis-deficient V79 Chinese hamster cell lines. Cancer Res. 1990;50:2713–2718. [PubMed] [Google Scholar]
- 67.Singh B., Gupta R.S. Mutagenic responses of thirteen anticancer drugs on mutation induction at multiple genetic loci and on sister chromatid exchanges in Chinese hamster ovary cells. Cancer Res. 1983;43:577–584. [PubMed] [Google Scholar]
- 68.Lambert S., Lopez B.S. Role of RAD51 in sister-chromatid exchanges in mammalian cells. Oncogene. 2001;20:6627–6631. doi: 10.1038/sj.onc.1204813. [DOI] [PubMed] [Google Scholar]
- 69.Dronkert M.L., Beverloo H.B., Johnson R.D., Hoeijmakers J.H., Jasin M., Kanaar R. Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol.Cell. Biol. 2000;20:3147–3156. doi: 10.1128/mcb.20.9.3147-3156.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsuoka A., Lundin C., Johansson F., Sahlin M., Fukuhara K., Sjoberg B.M., Jenssen D., Onfelt A. Correlation of sister chromatid exchange formation through homologous recombination with ribonucleotide reductase inhibition. Mutat. Res. 2004;547:101–107. doi: 10.1016/j.mrfmmm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 71.Tomimatsu N., Mukherjee B., Deland K., Kurimasa A., Bolderson E., Khanna K.K., Burma S. Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions. DNA Repair (Amst) 2012;11:441–448. doi: 10.1016/j.dnarep.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Langelier M.F., Planck J.L., Roy S., Pascal J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012;336:728–732. doi: 10.1126/science.1216338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eustermann S., Videler H., Yang J.C., Cole P.T., Gruszka D., Veprintsev D., Neuhaus D. The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger. J. Mol. Biol. 2011;407:149–170. doi: 10.1016/j.jmb.2011.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robert I., Karicheva O., Reina San Martin B., Schreiber V., Dantzer F. Functional aspects of PARylation in induced and programmed DNA repair processes: Preserving genome integrity and modulating physiological events. Mol. Aspects Med. 2013;34:1138–1152. doi: 10.1016/j.mam.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 75.De Vos M., Schreiber V., Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012;84:137–146. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 76.Lieber M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 77.Cheng Q., Barboule N., Frit P., Gomez D., Bombarde O., Couderc B., Ren G.S., Salles B., Calsou P. Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 2011;39:9605–9619. doi: 10.1093/nar/gkr656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Couto C.A., Wang H.Y., Green J.C., Kiely R., Siddaway R., Borer C., Pears C.J., Lakin N.D. PARP regulates nonhomologous end joining through retention of Ku at double-strand breaks. J. Cell. Biol. 2011;194:367–375. doi: 10.1083/jcb.201012132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grundy G.J., Rulten S.L., Zeng Z., Arribas-Bosacoma R., Iles N., Manley K., Oliver A., Caldecott K.W. APLF promotes the assembly and activity of non-homologous end joining protein complexes. Embo J. 2013;32:112–125. doi: 10.1038/emboj.2012.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shao Z., Davis A.J., Fattah K.R., So S., Sun J., Lee K.J., Harrison L., Yang J., Chen D.J. Persistently bound Ku at DNA ends attenuates DNA end resection and homologous recombination. DNA Repair (Amst) 2012;11:310–316. doi: 10.1016/j.dnarep.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu D., Topper L.M., Wilson T.E. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics. 2008;178:1237–1249. doi: 10.1534/genetics.107.083535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tang J., Cho N.W., Cui G., Manion E.M., Shanbhag N.M., Botuyan M.V., Mer G., Greenberg R.A. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013;20:317–325. doi: 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hartlerode A.J., Guan Y., Rajendran A., Ura K., Schotta G., Xie A., Shah J.V., Scully R. Impact of histone H4 lysine 20 methylation on 53BP1 responses to chromosomal double strand breaks. PLoS One. 2012;7 doi: 10.1371/journal.pone.0049211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lehtio L., Jemth A.S., Collins R., Loseva O., Johansson A., Markova N., Hammarstrom M., Flores A., Holmberg-Schiavone L., Weigelt J., et al. Structural basis for inhibitor specificity in human poly(ADP-ribose) polymerase-3. J. Med. Chem. 2009;52:3108–3111. doi: 10.1021/jm900052j. [DOI] [PubMed] [Google Scholar]
- 85.Loseva O., Jemth A.S., Bryant H.E., Schuler H., Lehtio L., Karlberg T., Helleday T. PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J. Biol. Chem. 2010;285:8054–8060. doi: 10.1074/jbc.M109.077834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miller K.M., Tjeertes J.V., Coates J., Legube G., Polo S.E., Britton S., Jackson S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010;17:1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ogiwara H., Ui A., Otsuka A., Satoh H., Yokomi I., Nakajima S., Yasui A., Yokota J., Kohno T. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011;30:2135–2146. doi: 10.1038/onc.2010.592. [DOI] [PubMed] [Google Scholar]
- 88.Vissers J.H., van Lohuizen M., Citterio E. The emerging role of Polycomb repressors in the response to DNA damage. J. Cell. Sci. 2012;125:3939–3948. doi: 10.1242/jcs.107375. [DOI] [PubMed] [Google Scholar]
- 89.Chou D.M., Adamson B., Dephoure N.E., Tan X., Nottke A.C., Hurov K.E., Gygi S.P., Colaiacovo M.P., Elledge S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. U. S. A. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ismail I.H., Gagne J.P., Caron M.C., McDonald D., Xu Z., Masson J.Y., Poirier G.G., Hendzel M.J. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res. 2012;40:5497–5510. doi: 10.1093/nar/gks222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gonzalez M.E., DuPrie M.L., Krueger H., Merajver S.D., Ventura A.C., Toy K.A., Kleer C.G. Histone methyltransferase EZH2 induces Akt-dependent genomic instability and BRCA1 inhibition in breast cancer. Cancer Res. 2011;71:2360–2370. doi: 10.1158/0008-5472.CAN-10-1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Polo S.E., Kaidi A., Baskcomb L., Galanty Y., Jackson S.P. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. Embo J. 2010;29:3130–3139. doi: 10.1038/emboj.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lindgren A.E., Karlberg T., Thorsell A.G., Hesse M., Spjut S., Ekblad T., Andersson C.D., Pinto A.F., Weigelt J., Hottiger M.O., et al. A PARP inhibitor with selectivity toward ADP-ribosyltransferase ARTD3/PARP3. ACS Chem. Biol. 2013;8:1698–1703. doi: 10.1021/cb4002014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.