


Abstract
Non-coding (nc)RNAs are important structural and regulatory molecules. Accurate determination of the primary sequence and secondary structure of ncRNAs is important for understanding their functions. During cDNA synthesis, RNA 3′ end stem-loops can self-prime reverse transcription, creating RNA–cDNA chimeras. We found that chimeric RNA–cDNA fragments can also be detected at 5′ end stem-loops, although at much lower frequency. Using the Gubler–Hoffman method, both types of chimeric fragments can be converted to cDNA during library construction, and they are readily detectable in high-throughput RNA sequencing (RNA-seq) experiments. Here, we show that these chimeric reads contain valuable information about the boundaries of ncRNAs. We developed a bioinformatic method, called Vicinal, to precisely map the ends of numerous fruitfly, mouse and human ncRNAs. Using this method, we analyzed chimeric reads from over 100 RNA-seq datasets, the results of which we make available for users to find RNAs of interest. In summary, we show that Vicinal is a useful tool for determination of the precise boundaries of uncharacterized ncRNAs, facilitating further structure/function studies.
INTRODUCTION
Non-coding RNAs (ncRNAs) are functional RNA molecules that are not translated into proteins. Many categories of ncRNAs have been discovered and characterized. These include RNAs that carry out basic cellular functions such as pre-mRNA splicing (small nuclear RNAs, snRNAs) and mRNA translation (tRNAs and rRNAs) (1). Also included are the small nucleolar (sno)RNAs and small Cajal body (sca)RNAs that guide post-transcriptional modification of rRNAs and snRNAs, respectively (1). Not only are ncRNA components of the core gene expression machinery, but they are also involved in multiple aspects of genetic regulation. This latter feature has been widely recognized with the discovery of microRNAs, siRNAs, piRNAs, lncRNAs, etc. (2). The regulatory activities of the ncRNAs include roles in chromatin remodeling, transcription, splicing, translation, RNA stability and even the stability and translocation of proteins (1–5). These functions usually depend upon their primary sequence and secondary structure in order to mediate interactions with proteins and other nucleic acids. Therefore, accurate determination of the RNA primary sequence is important for subsequent functional studies.
The rapid development in experimental and computational methodologies has significantly increased our ability to identify and study new ncRNAs. High-throughput sequencing of the transcriptome (RNA-seq) has been widely used for its high sensitivity and nucleotide resolution, and revealed hundreds to thousands of short and long ncRNAs in organisms from all three domains of life (6–8). De novo predictions based on evolutionary conservation and thermodynamic folding have also identified large numbers of ncRNAs and structured RNA elements in the genome (9,10). However, these methods do not provide enough resolution to accurately define the ends of the ncRNAs (11), and ends of the most ncRNAs are not well defined. Traditional methods of RNA end determination, such as 5′ RACE (Rapid Amplification of cDNA Ends) and 3′ RACE (rapid amplification of cDNA ends), although accurate, are labor-intensive and suffer from very low throughput (12,13). More advanced high-throughput experimental methods have been developed recently to map RNA ends, e.g. (14,15), but many of these methods are complicated and/or require the presence of poly(A) tails. In addition, new ways of analyzing the vast amount of existing RNA-seq data will be cost-effective and useful for gaining insights into various aspects of RNA structure and processing.
The traditional method for preparing cDNA libraries was developed by Gubler and Hoffman (16), which uses reverse transcriptase for first strand cDNA synthesis, RNase H, Escherichia coli DNA polymerase I and DNA ligase for second strand synthesis. This method is also commonly used for RNA-seq library preparation. Within certain RNA-seq datasets whose libraries were prepared using the Gubler–Hoffman method, we have discovered that a large number of the ‘unmappable’ reads are chimeric. That is, these reads consist of two parts: one from the 5′ or 3′ end of the RNA, and the other from an internal region of the RNA, on the opposite strand. This phenomenon clearly suggests self-priming from the 3′ end stem-loop, or ligation of the 5′ end stem-loop during cDNA library preparation. Using the chimeric reads from existing datasets, we developed a program, called Vicinal, to precisely determine the boundaries of ncRNAs and provide support for the predicted terminal stem-loops.
MATERIALS AND METHODS
Total RNA-seq of fruit fly larvae, pupae and pharate adults
Total RNA was extracted from third instar larvae, pupae and pharate adult flies and treated with DNase I to remove DNA contamination. Ribosomal RNAs were removed from the samples using the Ribo-Zero Human/Mouse/Rat kit (Epicentre). A TruSeq RNA Sample Preparation Kit v2 (Illumina) was used for barcoding, multiplexing and cDNA library preparation. The TruSeq procedure first fragments the rRNA-depleted samples and performs first strand synthesis using reverse transcriptase and random primers. The second strand synthesis uses DNA polymerase I and RNase H. The cDNA fragments then go through an end repair by adding a single adenosine at the ends. Adapters are ligated after repair. Paired end (2 × 48) sequencing was performed on an Illumina HiSeq 2000 platform. The data were deposited in the Gene Expression Omnibus with accession number GSE50711, and named Fly_larva3_48nt, Fly_pupa_48nt and Fly_pharate_48nt.
Additional RNA-seq datasets
Additional RNA-seq data used in this study were generated by our lab and others. They are listed as follows, together with their Short Read Archive accession numbers. Here we also briefly describe library preparation methods used to obtain these data, to help understand the Vicinal methodology. Fly_ovary_RIP_35nt: SRR120120-SRR120139 and SRR287104-SRR287107 (24 datasets) (17). Fly_S2_45nt: SRR345574-SRR345591 (18 datasets) (18). Mouse_ES_40nt: SRR392624-SRR392626 (3 datasets) (18). Mouse_ES_51nt: SRR915881-SRR915888 and SRR941123-SRR941140 (26 datasets) (19). Mouse_satellite_50nt: SRR953246 (1 dataset) (20). Human_HCT116_50nt: SRR901290-SRR901292 (3 datasets) (21).
The fly_ovary_RIP_35nt libraries were described previously (17). Briefly, Sm protein containing ribonucleoprotein (RNP) complexes from Drosophila ovaries were immunoprecipitated using anti-Sm or anti-GFP (green fluorescent protein) antibodies and the associated RNAs were purified. No polyA selection or rRNA removal was performed on the immunopurified RNA. First strand synthesis was carried out using a SuperScript III kit (Life Technologies). Second strand synthesis was performed using E. coli DNA polymerase I and RNase H (Life Technologies). Double-stranded cDNAs were made into libraries and sequenced using Illumina Genome Analyzer II. The fly_larva3_48nt, fly_pupa_48nt RNA-seq datasets were generated the same way as the fly_pharate_48nt described above. The Shilatifard lab generated the fly_S2_45nt, mouse_ES_40nt, mouse_ES_51nt and human_HCT_116_50nt RNA-seq datasets (18,19,21). Ribosomal RNA was removed from two micrograms of DNase-treated total RNA using the Ribo-Zero kit from Epicentre, and libraries were made using the Tru-seq mRNA kit from Illumina. The generation of mouse_satellite_50nt dataset by the Sartorelli lab also followed a similar protocol (20).
Bioinformatic pipeline
To obtain a rough estimate of the sequencing coverage and length of ncRNAs, the RNA-seq reads were mapped to reference genomes using Bowtie, allowing a maximum of two mismatches (end-to-end mapping) (22,23). Splicing was not considered in read mapping as only intronless ncRNAs were investigated in this study. Then the same raw reads were also mapped to genome references using Bowtie2 in the sensitive-local mode, which allows softclipping. The Bowtie- and Bowtie2-mapped reads were used to make bedgraph files for visualization in a genome browser. These bedgraph tracks were only shown in the analysis of snRNA:U1.
In order to identify self-priming and ligation events, the Bowtie2 mappable reads were filtered (using samsoftfilter.py, see the software package and instructions therein) to select reads that are only partially mapped to the genome, leaving at least n nucleotides from either end that are not mappable (n > 5). After filtering, the unmappable parts of the partially mappable reads were mapped again to the vicinity of the mappable parts (using Vicinal_1.0.py and Vicinal_2.0.py). This step generates a SAM (sequence alignment/map) file of chimeric reads and two wiggle files containing the coverage data for both the plus and minus strands. To make the method easy to use, we prepared a complete set of command line instructions. The scripts and instructions are available for download from the following website: https://sites.google.com/site/zhipeng0426/programming.
Implementation of the Vicinal algorithm
Samsoftfilter.py parses the CIGAR information from the input SAM file to find reads that have at least one softclipped region longer than a defined value (default n > 5). Shorter softclipped regions (n ≤ 5) could be sequencing errors or other kinds of chimeric sequences and therefore not considered in subsequent analysis. The softclipped reads were processed using Vicinal_1.0 and Vicinal_2.0.py scripts. The purpose of the Vicinal_1.0 and Vicinal_2.0.py script is to map the unmappable regions of the partially mapped reads to the vicinity of the mappable parts, on the opposite strand. We term this process ‘vicinal mapping’ to distinguish it from the Bowtie2 terminology of ‘local mapping’. Vicinal_1.0.py stores an initialized dictionary for fast processing but is only appropriate for genomes with smaller chromosomes, e.g. fly and nematode, whereas Vicinal_2.0.py does not store an initialized dictionary, and is slower, but can be used for genomes with larger chromosomes such as mouse and human. In order to map the softclipped reads efficiently and minimize memory footprint, the reads were sorted by chromosomal position. Once the reads mapped to one chromosome are processed, the results are written to output files (file_prefix_chim.sam for chimeric reads, file_prefix_1.wig and file_prefix_2.wig for coverage). Prior to vicinal mapping, the CIGAR code from the softclipped SAM file is parsed, and accordingly each read is divided into two or three parts, S+M, M+S or S+M+S, depending on whether there are softclipped fragments on the 5′ and/or 3′ end, where S represents 'softclipped' and M represents 'matched.' Internal mismatches were ignored. A region around each mapped fragment (with a radius defined by the users, default is 100 nt) was extracted from the reference genome on the opposite strand. Then the softclipped fragments were searched against the extracted region and a total of one match is allowed. Once a match is found, the record for that read is output to a SAM file, and the coordinates of the mapped fragments on both strands were calculated and output to the two wiggle files. The wiggle files can be further converted to smaller gzipped bedgragh files for efficient storage and transfer. See detailed instructions in the Vicinal software package: https://sites.google.com/site/zhipeng0426/programming.
ncRNA lists and generation of lists with chimeric read numbers
Lists of fruit fly, mouse and human ncRNA coordinates used in the analysis were generated as follows. The Drosophila ncRNA list was downloaded from UCSC Genome Bioinformatics site (http://hgdownload.soe.ucsc.edu/goldenPath/dm3/database/), matched with gene names from Flybase (http://flybase.org/static_pages/downloads/COORD.html) and rearranged according to the format described in the Vicinal software. The mouse ncRNA list was downloaded from the mouse genome informatics (MGI) database at Jackson Laboratory (ftp://ftp.informatics.jax.org/pub/reports/MGI_MRK_Coord.rpt). The human ncRNA list was downloaded from Ensembl (ftp://ftp.ensembl.org/pub/release-74/fasta/homo_sapiens/ncrna/) and rearranged accordingly. The number of chimeric reads for each ncRNA can be obtained using the readnum.sh script, which depends on the samtools package (24).
Northern blotting
S2 cells were homogenized in TRIzol (Life Technologies) and total RNA was extracted following manufacturer's instructions. RNA was electrophoresed in 4–12% TBE-Urea polyacrylamide gels (Life Technologies), transferred to nylon membranes and probed with 32P-labeled polymerase chain reaction (PCR) products corresponding to the Drosophila melanogaster U2 and LU snRNA cDNAs.
RNA secondary structure prediction
The secondary structures of non-coding RNAs were predicted using either UNAfold or the Vienna RNA Package with default parameter settings (25,26). Alternative secondary structures (conformers) were occasionally adjusted manually to fit the chimeric reads. Structured alignments of ncRNAs were performed using LocARNA (global standard alignment) (27). Secondary structures of the predicted RNAs were drawn using VARNA (28).
RESULTS
Previously, we carried out an RNA-immunoprecipitation sequencing (RIP-seq) analysis to identify RNAs that co-purify with Sm proteins in Drosophila and human cells (17). During preparation of the sequencing libraries, we used either oligo-dT or random hexamer primers for first strand cDNA synthesis from RNA. Curiously, we found that both random and oligo-dT primed libraries contained large numbers of snRNA transcripts. This latter result was unexpected because snRNAs are not polyadenylated. To explain this observation, we considered the possibility that the snRNA reads detected in the oligo-dT primed libraries might be oligoadenylated RNA degradation intermediates (29). However, manual inspection of reads derived from snRNA 3′ ends did not reveal oligo(A) extensions in either the oligo-dT or random hexamer primed libraries; instead, the (non-templated) extensions appear to be products of self-priming from stem-loop sequences that are typically present at the 3′ ends of snRNAs. Though much less frequent, we also found reads that contain 5′ extensions, which might be the result of ligation events between cDNA and RNA 5′ stem-loops. A diagram of possible mechanisms for the generation of 5′ end and 3′ end chimeric reads is presented in Figure 1. The 3′ end stem-loop can serve as a primer for first strand cDNA synthesis. cDNA fragments that are close to the 5′ end stem-loop can be ligated with the 5′ end RNA by DNA ligase. The two types of DNA–RNA chimera could, in principle, serve as templates for second strand synthesis. The resultant double-stranded DNA could then be further ligated with adapters and sequenced. The 5′ cap structures present at many ncRNAs provide one explanation for the low efficiency of the 5′ ligation. However, it is not entirely clear how DNA polymerase I uses DNA–RNA chimeras as templates for second strand synthesis. The diagram in Figure 1 is provided for illustrative purposes to help understand how chimeric reads might be generated.
Figure 1.
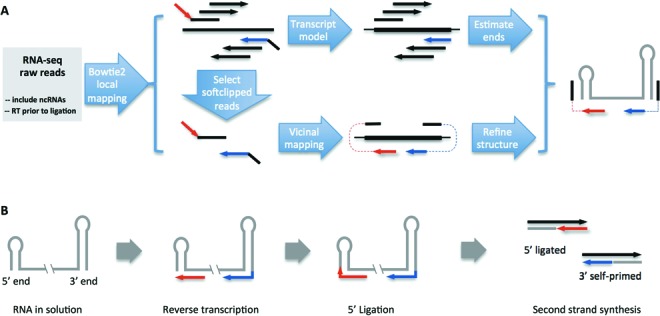
Vicinal pipeline and possible mechanisms for the generation of chimeric reads. (A) Flowchart of the analysis pipeline. The RNA-seq libraries are prepared such that they do not exclude ncRNAs, and the reverse transcription step precedes adapter ligation. The RNA-seq reads are first aligned to the genome using Bowtie2 in the local-mapping mode (--sensitive-local). Then partially mapped reads are selected and vicinally mapped using Vicinal. Bowtie2 mapped reads are directly used to roughly estimate the boundaries of the ncRNAs, while the Vicinal mapped reads are used to determine the boundaries. Finally, the secondary structure is predicted and adjusted to fit the chimeric reads. The convention of colors is consistent throughout all the figures. (B) Many ncRNAs in solution adopt secondary structures with terminal stem-loops. During reverse transcription, the 3′ end stem-loop can serve as a primer, in addition to primers added to the solution, for cDNA synthesis (red and blue lines with arrows are cDNA fragments). After cDNA synthesis, the cDNA fragments close to the 5′ end stem-loop can be ligated to the stem-loop. The 5′ end and 3′ end cDNA chimeras can further serve as templates for second strand DNA synthesis, thus producing cDNA fragments for subsequent adapter ligation and deep sequencing.
The Vicinal algorithm
Initial examination of the chimeric reads derived from snRNAs reveals two important features, irrespective of whether they arose via self-priming or ligation. First, the two parts of each chimeric read usually map close to one another, within 100 nt. This distance is basically determined by the size of the terminal stem-loop. Second, the two parts of each chimera map to opposite strands of the encoding DNA, unlike reads derived from spliced RNAs, which map to the same strand. Based on these properties, we developed an analysis pipeline to identify reads that are derived from self-priming and ligation events (Figure 1).
For vicinal mapping to work, the RNA-seq libraries must be prepared in a way that allows for self-priming. This is usually accomplished by cDNA synthesis prior to adapter ligation, on RNA samples that contain ncRNAs (Figure 1A). Because chimeric reads represent only a small portion of the total number of reads (see read mapping statistics in Table 1, and Figure 2A and B for an example), efficient processing of raw RNA-seq data is important for subsequent analysis. We used Bowtie2 for preliminary mapping, because it is fast and allows softclipping of the reads for local (partial) mapping (23). The Bowtie2 mapping results provide a rough estimate of the coverage and size of transcripts. For the locally (partially) mapped reads, only the longer segment of the read is mapped to the genome, whereas the shorter segment is softclipped/ignored.
Table 1. RNA-seq datasets used in the study.
| Sample | Reference | Read length | Mappable | Softclipped | Chimeric | % Chimeric |
|---|---|---|---|---|---|---|
| fly_ovary_RIP | Lu et al. (2014) (17) | 35 | 87594638 | 11334332 | 990969 | 1.13 |
| fly_pharate | This study | 48 | 124544603 | 13919998 | 226981 | 0.18 |
| fly_S2 | Smith et al. (2011) (18) | 45/50 | 224898608 | 18788217 | 387406 | 0.17 |
| fly_larva3 | This study | 48 | 241674561 | 25221457 | 1294206 | 0.53 |
| fly_pupa | This study | 48 | 129388545 | 12737427 | 182526 | 0.14 |
| mouse_ES | Smith et al. (2011) (18) | 40 | 101615022 | 5573492 | 93137 | 0.09 |
| mouse_ES | Hu et al. (2013) (19) | 51 | 712409456 | 53374271 | 438910 | 0.06 |
| mouse_satellite | Mousavi et al. (2013) (20) | 50 | 41019420 | 1910902 | 57034 | 0.14 |
| human_HCT116 | Hu et al. (2013) (21) | 50 | 90190285 | 6695509 | 123856 | 0.14 |
Figure 2.

Vicinal analysis of known fly ncRNAs, snRNA:U1 (A–E) and RNaseP:RNA (F–L). (A) Five genome browser tracks for snRNA:U1:95Cc are shown from the analysis of fly_ovary_RIP_35nt datasets: bt1_E2E (Bowtie1 end-to-end mapping of raw reads), bt2_local (Bowtie2 local mapping), soft (selecting softclipped reads from bt2_local), plus and minus (reads mappable by Vicinal). The thick blue bar represents the mature U1:95Cc transcript region. The vertical dashed blue lines align the 5′ and 3′ ends of the U1 transcript. Note that there are five functional U1 snRNA genes in the fly genome and only one is shown here. The high peaks mapped to the middle of U1 are artifacts from mapping short softclipped parts of reads. (B) Filtering reads from fly_ovary_35nt_plus/minus for reads close to the estimated U1 snRNA ends showed clear terminal pileup of reads. Reads for all five U1 snRNA paralogs were combined. Note the difference in scale for the 5′ end and 3′ end chimeric reads. (C) Detailed analysis of the chimeric reads that map to both ends. The first line is the genomic DNA sequences around the 5′ and 3′ ends. Subsequent lines are the manually aligned chimeric reads, where black letters represent parts mapped to the ends of the transcript, blue letters represent 3′ extensions mapped to the internal region on the opposite strand, and the red letters represent 5′ extensions mapped to the internal region on the opposite strand. The numbers before each chimeric read sequence are read counts. Note the differences between the extended genomic DNA and the terminal extensions in the chimeric reads. Only the top 10 groups of distinct reads are shown for the 3′ end, and top 6 groups of distinct reads for the 5′ end. (D) Predicted secondary structure that explains the production of the chimeric reads. The black lines represent parts of reads mapped to the ends of U1 snRNA, whereas the red and blue lines represent terminal extensions mapped to the internal regions of U1 snRNA. (E) Potential equilibrium in solution between the chimera-generating secondary structure (on the left, the same as in D) and the well-known physiological secondary structure in U1 snRNP (on the right). The normal secondary structure is unlikely to give rise to 5′ end ligated reads due to the long 5′ overhang. (F) Ten genome browser tracks for RNaseP:RNA are shown from the Vicinal analysis of five groups of fly RNA-seq data. For simplicity, the end-to-end mapping, local mapping and softclipped read tracks are not shown. Note the terminally adjusted read pileups; they are not filtered as in B for U1 snRNA. The 5′ end of the RNaseP:RNA is on the right. Chimeric reads were combined from all five groups of RNA-seq data, for subsequent detailed analysis. (G) Detailed analysis of the chimeric reads that map to 3′ end of RNaseP:RNA. (H) The chimera-generating secondary structure of the 3′ end of RNaseP:RNA. (I and K) Detailed analysis of reads mapped the 5′ end reveals two possible 5′ ends that differ by four nucleotides. (J and L) The chimera-generating secondary structures of the 5′ end of RNaseP:RNA. Note, the secondary structures shown here for the 5′ end and 3′ end are different from the physiological secondary structure of the RNaseP RNP.
After the initial mapping step, we filtered the mapped reads to select those with at least one softclipped segment longer than a defined size (e.g. 5 nt). The size of the softclipped segment is chosen so that the fragment can be uniquely mapped in the vicinity of the Bowtie2 mapped part of the read. The softclipped segments are then mapped ‘vicinally’, that is, mapped to a region within a certain distance (e.g. 100 nt) from the mapped segment, on the opposite strand (Figure 1A).
Once both segments are mapped, the junction is used to define the ends of the ncRNA, and terminal stem-loops in the predicted secondary structures are used to explain the source of the chimeric reads (Figure 1A and B). However, the presence of self-primed and ligated reads does not imply that the chimera-generating terminal stem-loops are stable in vivo. It is only evidence for the presence of the terminal stem-loops in solution, likely in equilibrium with other conformations (see Figures 2H and 3E, H, J and L). We generated lists of ncRNA genomic coordinates and used them to intersect with chimeric reads generated by Vicinal to make lists of ncRNAs with numbers of chimeric reads (see instructions and the results: https://sites.google.com/site/zhipeng0426/programming).
We have analyzed hundreds of RNA-seq datasets using Vicinal and found 115 of them containing self-primed and ligated chimeric reads. These datasets were sorted into nine different categories, according to organism, tissue and/or read length (Table 1). The data were generated by several different laboratories, demonstrating that such chimeric reads are not specific artifacts of a single lab. Five of the nine categories are sourced from the fruitfly, three from the mouse and one from human. Statistics of the Bowtie2 local mapping, filtering and Vicinal mapping are presented. Although the fraction of chimeric reads is not high in any of the datasets, there are enough of them to determine the ends of many ncRNAs. Given the large number of starting raw reads in most RNA-seq experiments, Vicinal analysis provides users with numerous ncRNAs with sufficient chimeric read coverage. These include snRNAs, snoRNAs, scaRNAs, 5.8S rRNA, 7SK RNA, 7SL RNA, RNaseP RNA, RNaseMRP RNA, etc. (see Figures 2 and 3, Supplementary Figures S1 and S2 and ncRNA lists from our website: https://sites.google.com/site/zhipeng0426/programming). The chimeric read coverage for these ncRNAs varies greatly from dozens to thousands of reads per RNA. However, certain types of ncRNAs, e.g. lncRNAs, miRNAs, siRNAs, etc., do not typically have chimeric reads and thus the Vicinal program is not applicable. These results demonstrate the utility of our approach in the analysis of multiple categories of ncRNAs, with a wide range of expression levels, in different organisms. Here we show several examples of using Vicinal to analyze several known and newly discovered fly ncRNAs. More examples of Vicinal analysis on fly, mouse and human ncRNAs are presented in Supplementary Figures S1 and S2.
Figure 3.
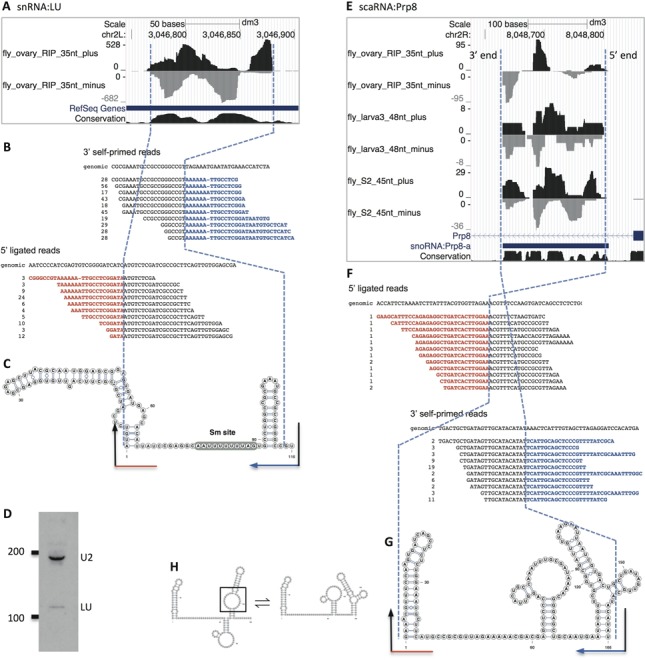
Vicinal analysis of snRNA:Like-U (LU) (A–D) and scaRNA:Prp8 (E–G). Please refer to Figure 2 for general description of the analysis flow. (A) Only two genome browser tracks for snRNA:LU are displayed: plus and minus, whereas the end-to-end mapping, local-mapping and softclipped read tracks are not shown. Note the size predicted by Jung et al. (32) (transcript model, the thick blue line) is longer than the size determined by Vicinal analysis. (B) Detailed analysis of the chimeric reads. Only the top 10 groups of distinct reads are shown. The 3′ ligated reads included variants, because sequencing the long stretch of adenosines unavoidably introduces errors. (C) The chimera-generating secondary structure for snRNA:LU. The Sm site is shown on the predicted secondary structure. (D) Northern blot of Drosophila U2 and LU snRNAs. (E) Six genome browser tracks for scaRNA:Prp8 are shown from the analysis of three groups of RNA-seq datasets, where chimeric reads for this RNA are available. Note that the earliest annotation labeled this ncRNA as a snoRNA (as shown in the gene annotation track); however, subsequent research suggests that it is a scaRNA. (F) Detailed analysis of the chimeric reads. (G) The chimera-generating secondary structure for scaRNA:Prp8. (H) Potential equilibrium between the more likely physiological secondary structure (on the left, with the pseudouridylation pocket open in the last stem-loop in a black box) and the chimera-generating secondary structure (on the right).
Confirmation of known snRNA:U1 and RNaseP:RNA ends using Vicinal
Most snRNAs have stem-loops at their 5′ and 3′ ends with very short overhangs, and their sequences and secondary structures are well characterized. Sm protein immunoprecipitations enrich for snRNAs, and among them, U1 is the most abundant. Therefore, we first analyzed chimeric reads derived from U1 snRNA as a proof-of-principle (Figure 2A–E). The chimeric reads for U1 are most abundant in the fly_ovary_RIP_35nt sample and therefore we only showed this group of RNA-seq data in the Vicinal analysis of U1.
The read coverage patterns for U1 snRNA, using Bowtie (end-to-end mapping) and Bowtie2 (local mapping), are not uniform (Figure 2A, and for other ncRNAs, data not shown). This is especially true near the two ends, because untemplated extensions in the reads are not mappable, and the priming and sequencing efficiency along U1 varies according to sequence and structural contexts. The non-uniformity of the read coverage makes estimation of transcript size difficult. However, selection of the Bowtie2 locally (partially) mapped reads clearly shows that many of the mappable fragments are justified to the left or right ends, suggesting the existence of softclipping in Bowtie2 mapping (Figure 2A and B). Vicinal mapping of the chimeric reads places the unmapped segments on the opposite strands (Figure 2A and B) and the terminally mapped half-reads indicate the presence of terminal stem-loops (Figure 2C). Patterns of read coverage showed clear end justification (Figure 2A and B, the dashed blue line, see also examples in subsequent figures).
Detailed alignment of the partially mapped U1 reads showed clear signs of chimera formation (Figure 2C). The presence of terminal overhangs and imperfect complementarity in the stem allows for definition of the boundaries. Importantly, the abundance of the 3′ end-derived chimeric reads confirmed the stable 3′ end stem-loop which allows for efficient self-priming, despite the presence of imperfect complementarity. In fact, the presence of base pair mismatches made it possible to define the ends with near single-nucleotide resolution. The identity of the few additional nucleotides (usually 1–2 nucleotides) close to the end of the mature transcript may interfere with the accuracy of end determination, but most of the time, they are short enough to allow near-nucleotide determination. In contrast to the 3′ end reads, there were many fewer reads derived from the U1 5′ end. The relative dearth of 5′ end chimeric reads is likely due to the fact that the 5′ overhang in the predicted secondary structure is quite long (∼11 nt) and that ligation to first strand cDNA is likely to be very inefficient, due to the presence of the trimethylguanosine (TMG) cap. Because cDNA library construction takes place on purified RNAs and not on stable RNPs, U1 may well adopt alternative secondary structures in solution. One such alternative U1 structural isomer (see Figure 2E) has no overhang and might be a better substrate for generation of chimeric 5′ end reads. Irrespective of the mechanism, the structure of the observed U1 chimeric reads is consistent with the known 5′ and 3′ ends of U1 snRNA (30).
RNaseP:RNA is a ribozyme that cleaves pre-tRNA 5′ end leader sequences during tRNA biogenesis and is an essential RNA in all life forms. Here we present a detailed analysis of RNaseP:RNA ends using Vicinal (Figure 2F–L). Chimeric reads are detectable for RNaseP:RNA in all five categories of fly RNA-seq data, with varied abundance (Figure 2F). We combined chimeric reads from all five of these groups, and Vicinal analysis revealed clear terminally justified chimeric reads for both 5′ and 3′ ends. The 3′ end chimeric reads clearly define a single end, with a maximum of two ambiguous nucleotides (Figure 2G and H), consistent with previous report (31). The 5′ ligated reads suggest two possible ends, differing by 4 nt (Figure 2I–L). One of the two 5′ ends is consistent with previous reports (Figure 2I and J) (31). It is likely that the other one (Figure 2K and L) represents a transcript that uses a different transcription start site or is subject to alternative 5′ processing. The terminal stem-loops that explain generation of 5′ and 3′ chimeric reads are different from the physiological secondary structure (present in the RNaseP RNP particle), but nonetheless they are very likely to exist in solution (31). Taken together, our method defines the boundaries of two known ncRNAs.
Vicinal analysis defines boundaries of newly discovered snRNAs and sno/scaRNAs
In order to show the utility of Vicinal in analysis of novel or under-studied ncRNAs, we have examined all Drosophila, mouse and human ncRNAs using Vicinal and detected chimeric reads in many of them. Here we show two ncRNAs that we discovered in our previous RIP-seq analysis (17). Dozens of additional examples are presented in Supplementary Figures S1 and S2.
snRNA:LU. Like-U is a newly evolved Sm-class snRNA (CR43708), present only in Drosophilid genomes (17). Vicinal analysis of LU snRNA revealed hundreds of terminally justified fragments and internally mapped second fragments at both the 5′ end and 3′ end of the transcript (Figure 3A). We analyzed these chimeric reads and predicted that the length of LU snRNA is 116 nt. The predicted secondary structure is shown in Figure 3C. Previously, Jung et al. (2010) analyzed publicly available RNA-seq data and identified a transcript from this locus, estimating its length to be 150–160 nt (32). To resolve this difference in length prediction, we performed northern blotting of LU snRNA, which showed a size that is consistent with our Vicinal analysis (110–120 nt, Figure 3D). This size is also consistent with the sequence conservation of LU orthologs among Drosophilids (17).
scaRNA:Prp8. Another novel Sm-associated ncRNA we discovered in our RIP-seq study is scaRNA:Prp8 (CR43600; Figure 4C). A previous transcriptomic study estimated its size to be 178 nt (8). Here, Vicinal analysis of three groups of RNA-seq data revealed dozens of terminally justified fragments and predicts a length of 168 nt (Figure 3E–H). This size is also consistent with the alignment of scaRNA orthologs in insects. The chimeric reads can be explained by an alternative conformation of the secondary structure (Figure 3G and H, right side). The other conformation (Figure 3H, left side) is likely to be the physiological one, due to the presence of an open pseudouridylation pocket (Figure 3H, black box), as reported recently by Deryusheva and Gall (33).
The determination of ncRNA ends following Vicinal mapping requires manual alignment of the chimeric reads and fitting onto predicted secondary structures. In order to make best use of the large amounts of chimeric reads mapped using Vicinal from Drosophila, mouse and human RNA-seq datasets, we provide them as lists for users to identify ncRNAs of their interest. The lists contain ncRNA identifiers and numbers of chimeric reads mapped to each ncRNA. Chimeric read coverage patterns can be visualized by importing the bedgraph track files into genome browsers. The chimeric reads for each ncRNA can be extracted from the Vicinal mapped BAM (binary alignment/map) files and manually aligned. In the future, additional RNA-seq datasets can be added to increase the chimeric read coverage on ncRNAs in the species analyzed, and potentially in other species as well.
DISCUSSION
In this study, we present a new bioinformatic tool, called Vicinal, to define the ends of ncRNAs with terminal stem-loops. This method takes advantage of the self-priming and ligation property of ncRNA 3′ and 5′ terminal stem-loops during library preparation using the Gubler–Hoffman method (16), and the power of massively parallel sequencing. Using Vicinal, we confirmed the boundaries of previously studied ncRNAs and also defined the boundaries of newly discovered ncRNAs from many different RNA-seq datasets from various species.
Although other methods are available for the determination of ncRNA ends, many of them are labor-intensive and require more experiments. Our analysis method makes use of published RNA-seq data and is cost-effective. More accurate determination of ends for more ncRNAs will be available with the publication of ever increasing amount RNA-seq data.
It has long been known that 3′ end self-priming of U3 snoRNA mediates pseudogene formation during the process of retrotransposition (34). Pseudogenes derived from other highly structured ncRNAs, including U1 and U2 snRNAs, are also known to form in this manner. Furthermore, self-priming from 3′ end stem-loops is a relatively common feature among certain single-stranded RNA and DNA viruses (35–37). This self-priming ability is required for proper replication of the viral genome. Moreover, the high efficiency and specificity of self-priming from terminal stem-loops has been exploited for quantification of small RNA levels by reverse transcriptase-polymerase chain reaction (RT-PCR), wherein a stem-loop RT primer is used instead of a conventional, unstructured primer (38). In contrast to the widespread use of 3′ self-priming in nature, we are not aware of previous findings regarding the phenomenon of ligation to 5′ end RNA stem-loops during cDNA library construction, and further studies will be needed in order to understand the mechanism.
The use of soft-clipped reads for mapping inevitably creates artifacts. Vicinal mapping sometimes assigns reads to exon–exon junctions, due to their short length after clipping (data not shown). Other kinds of artifacts are also observed, mainly in highly expressed ncRNAs, such as ribosomal RNAs and certain snRNAs (Figure 2A). However, such artifacts can be clearly distinguished from chimeras that are generated by self-priming and ligation. The latter have distinct features, such as terminally justified pileups. We note that certain RNA secondary structures are likely to be more favorable for chimera formation than others. Although such structures are not necessarily the most stable ones in solution or in vivo (see Figures 2D, H, J, L and 3G), the boundary mapping procedure described here can easily pick up such low-frequency priming events.
In summary, the method described above enables highly sensitive analysis of ncRNA boundaries. The use of fast short-read mappers (we used Bowtie2) in combination with rapid local alignment of what would otherwise be considered ‘unmappable’ fragments allows for efficient processing of large datasets in a relatively short period of time. Because terminal stem-loops and internal single-stranded regions are common features of many ncRNAs, our method should prove useful for a wide variety of studies in RNA biology.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank the members of the Matera laboratory for numerous helpful comments and suggestions. We also thank Dr Xiaojun Guan (Sequenom, San Diego, CA) and Liang Shan (UNC Department of Computer Science) for advice and help with computer programming.
FUNDING
National Institutes of Health (NIH) [R01-GM053034 to A.G.M.]. Source of open access funding: NIH [R01-GM053034].
Conflict of interest statement None declared.
REFERENCES
- 1.Matera A.G., Terns R.M., Terns M.P. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Bio. 2007;8:209–220. doi: 10.1038/nrm2124. [DOI] [PubMed] [Google Scholar]
- 2.Mattick J.S., Makunin I.V. Non-coding RNA. Hum. Mol. Genet. 2006:R17–29. doi: 10.1093/hmg/ddl046. 15. [DOI] [PubMed] [Google Scholar]
- 3.Kondrashov A.V., Kiefmann M., Ebnet K., Khanam T., Muddashetty R.S., Brosius J. Inhibitory effect of naked neural BC1 RNA or BC200 RNA on eukaryotic in vitro translation systems is reversed by poly(A)-binding protein (PABP) J. Mol. Biol. 2005;353:88–103. doi: 10.1016/j.jmb.2005.07.049. [DOI] [PubMed] [Google Scholar]
- 4.Yang Z., Zhu Q., Luo K., Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- 5.Walter P., Blobel G. Signal recognition particle contains a 7S RNA essential for protein translocation across the endoplasmic reticulum. Nature. 1982;299:691–698. doi: 10.1038/299691a0. [DOI] [PubMed] [Google Scholar]
- 6.Croucher N.J., Thomson N.R. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010;13:619–624. doi: 10.1016/j.mib.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graveley B.R., Brooks A.N., Carlson J.W., Duff M.O., Landolin J.M., Yang L., Artieri C.G., van Baren M.J., Boley N., Booth B.W., et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Washietl S., Hofacker I.L., Stadler P.F. Fast and reliable prediction of noncoding RNAs. Proc. Natl. Acad. Sci. U.S.A. 2005;102:2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pedersen J.S., Bejerano G., Siepel A., Rosenbloom K., Lindblad-Toh K., Lander E.S., Kent J., Miller W., Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput. Biol. 2006;2:e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Will S., Joshi T., Hofacker I.L., Stadler P.F., Backofen R. LocARNA-P: accurate boundary prediction and improved detection of structural RNAs. RNA. 2012;18:900–914. doi: 10.1261/rna.029041.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scotto-Lavino E., Du G., Frohman M.A. 3′ end cDNA amplification using classic RACE. Nat. Protoc. 2006;1:2742–2745. doi: 10.1038/nprot.2006.481. [DOI] [PubMed] [Google Scholar]
- 13.Scotto-Lavino E., Du G., Frohman M.A. 5′ end cDNA amplification using classic RACE. Nat. Protoc. 2006;1:2555–2562. doi: 10.1038/nprot.2006.480. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi H., Lassmann T., Murata M., Carninci P. 5′ end-centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nat. Protoc. 2012;7:542–561. doi: 10.1038/nprot.2012.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruan X., Ruan Y. Genome wide full-length transcript analysis using 5′ and 3′ paired-end-tag next generation sequencing (RNA-PET) Methods Mol. Biol. 2012;809:535–562. doi: 10.1007/978-1-61779-376-9_35. [DOI] [PubMed] [Google Scholar]
- 16.Gubler U., Hoffman B.J. A simple and very efficient method for generating cDNA libraries. Gene. 1983;25:263–269. doi: 10.1016/0378-1119(83)90230-5. [DOI] [PubMed] [Google Scholar]
- 17.Lu Z., Guan X., Schmidt C.A., Matera A.G. RIP-seq analysis of eukaryotic Sm proteins identifies three major categories of Sm-containing ribonucleoproteins. Genome Biol. 2014;15:R7. doi: 10.1186/gb-2014-15-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith E.R., Lin C., Garrett A.S., Thornton J., Mohaghegh N., Hu D., Jackson J., Saraf A., Swanson S.K., Seidel C., et al. The little elongation complex regulates small nuclear RNA transcription. Mol. Cell. 2011;44:954–965. doi: 10.1016/j.molcel.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu D., Garruss A.S., Gao X., Morgan M.A., Cook M., Smith E.R., Shilatifard A. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1093–1097. doi: 10.1038/nsmb.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mousavi K., Zare H., Dell’orso S., Grontved L., Gutierrez-Cruz G., Derfoul A., Hager G.L., Sartorelli V. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell. 2013;51:606–617. doi: 10.1016/j.molcel.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu D., Smith E.R., Garruss A.S., Mohaghegh N., Varberg J.M., Lin C., Jackson J., Gao X., Saraf A., Florens L., et al. The little elongation complex functions at initiation and elongation phases of snRNA gene transcription. Mol. Cell. 2013;51:493–505. doi: 10.1016/j.molcel.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markham N.R., Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol. Biol. 2008;453:3–31. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- 26.Hofacker I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Will S., Reiche K., Hofacker I.L., Stadler P.F., Backofen R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007;3:e65. doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darty K., Denise A., Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura R., Takeuchi R., Takata K., Shimanouchi K., Abe Y., Kanai Y., Ruike T., Ihara A., Sakaguchi K. TRF4 is involved in polyadenylation of snRNAs in Drosophila melanogaster. Mol. Cell. Biol. 2008;28:6620–6631. doi: 10.1128/MCB.00448-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lo P.C., Mount S.M. Drosophila melanogaster genes for U1 snRNA variants and their expression during development. Nucleic Acids Res. 1990;18:6971–6979. doi: 10.1093/nar/18.23.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marquez S.M., Harris J.K., Kelley S.T., Brown J.W., Dawson S.C., Roberts E.C., Pace N.R. Structural implications of novel diversity in eucaryal RNase P RNA. RNA. 2005;11:739–751. doi: 10.1261/rna.7211705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung C.H., Hansen M.A., Makunin I.V., Korbie D.J., Mattick J.S. Identification of novel non-coding RNAs using profiles of short sequence reads from next generation sequencing data. BMC Genomics. 2010;11:77. doi: 10.1186/1471-2164-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deryusheva S., Gall J.G. Novel small Cajal-body-specific RNAs identified in Drosophila: probing guide RNA function. RNA. 2013;19:1802–1814. doi: 10.1261/rna.042028.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernstein L.B., Mount S.M., Weiner A.M. Pseudogenes for human small nuclear RNA U3 appear to arise by integration of self-primed reverse transcripts of the RNA into new chromosomal sites. Cell. 1983;32:461–472. doi: 10.1016/0092-8674(83)90466-x. [DOI] [PubMed] [Google Scholar]
- 35.Salzman L.A., Fabisch P. Nucleotide sequence of the self-priming 3′ terminus of the single-stranded DNA extracted from the parvovirus Kilham rat virus. J. Virol. 1979;30:946–950. doi: 10.1128/jvi.30.3.946-950.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bourguignon G.J., Tattersall P.J., Ward D.C. DNA of minute virus of mice: self-priming, nonpermuted, single-stranded genome with a 5′-terminal hairpin duplex. J. Virol. 1976;20:290–306. doi: 10.1128/jvi.20.1.290-306.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tuiskunen A., Leparc-Goffart I., Boubis L., Monteil V., Klingstrom J., Tolou H.J., Lundkvist A., Plumet S. Self-priming of reverse transcriptase impairs strand-specific detection of dengue virus RNA. J. Gen. Virol. 2010;91:1019–1027. doi: 10.1099/vir.0.016667-0. [DOI] [PubMed] [Google Scholar]
- 38.Chen C., Ridzon D.A., Broomer A.J., Zhou Z., Lee D.H., Nguyen J.T., Barbisin M., Xu N.L., Mahuvakar V.R., Andersen M.R., et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.