


Abstract
Accumulating evidence suggests that dormant DNA replication origins play an important role in the recovery of stalled forks. However, their functional interactions with other fork recovery mechanisms have not been tested. We previously reported intrinsic activation of the Fanconi anemia (FA) pathway in a tumor-prone mouse model (Mcm4chaos3) with a 60% loss of dormant origins. To understand this further, we introduced a null allele of Fancc (Fancc−), encoding a member of the FA core complex, into the Mcm4chaos3 background. Primary embryonic fibroblasts double homozygous for Mcm4chaos3 and Fancc− (Mcm4chaos3/chaos3;Fancc−/−) showed significantly increased levels of markers of stalled/collapsed forks compared to either single homozygote. Interestingly, a loss of dormant origins also increased the number of sites in which replication was delayed until prophase, regardless of FA pathway activation. These replication defects coincided with substantially elevated levels of genome instability in Mcm4chaos3/chaos3;Fancc−/− cells, resulting in a high rate of perinatal lethality of Mcm4chaos3/chaos3;Fancc−/− mice and the accelerated tumorigenesis of surviving mice. Together, these findings uncover a specialized role of dormant origins in replication completion while also identifying important functional overlaps between dormant origins and the FA pathway in maintaining fork progression, genome stability, normal development and tumor suppression.
INTRODUCTION
Origin licensing builds a fundamental basis for genome stability during eukaryotic DNA replication as it provides replication origins with the competency to fire and restricts their firing to once per S phase (1–3). This process occurs during the late M to early G1 phases of the cell cycle, when heterohexameric complexes of the minichromosome maintenance proteins (MCM2-7), essential components of the replicative helicase, are loaded onto chromatin (4–6). While any genomic loci bound by MCM2-7 complexes can potentially act as origins, only a small fraction of them (∼10%) assemble active helicases with their co-factors to unwind the DNA and initiate genome duplication in S phase (7,8). In fact, chromatin-bound MCM2-7 complexes exist in a large excess (10- to 20-fold) over the number of replication origins that actually fire in S phase (9–12), thereby licensing additional origins termed dormant origins. Although dormant origins represent the vast majority (>90%) of all licensed origins (13,14), their role in DNA replication has only recently been revealed. These dormant origins can be activated as ‘backups’ under conditions of replication stress to compensate for slow fork progression and rescue stalled replication forks, thereby contributing to completion of DNA replication (13–15).
Using a mouse model called Mcm4chaos3, we demonstrated that dormant origins also play an important role in the rescue of stalled forks even in unchallenged S phase (16). Mcm4chaos3 is a hypomorphic allele encoding a Phe345Ile change in the MCM4 protein, a subunit of the MCM2-7 complex (17). Cells homozygous for this allele (Mcm4chaos3/chaos3) exhibit a decreased rate of assembly of the MCM2-7 complex, leading to an ∼60% loss of chromatin-bound MCM2-7 complexes (16). This results in a significant reduction of dormant origins, causing the accumulation of stalled forks and increased levels of spontaneous micronuclei (MN) in Mcm4chaos3/chaos3 cells. Reflecting intrinsic genome instability, Mcm4chaos3/chaos3 mice are highly prone to spontaneous tumors (16,17). These properties of Mcm4chaos3/chaos3 mice substantiate the formerly underappreciated role of dormant origins in stalled fork recovery (18). In our previous work (16), Mcm4chaos3/chaos3 cells were also found to exhibit intrinsic activation of the Fanconi anemia (FA) pathway of DNA repair, though the functional relevance of this had yet to be determined.
FA is a rare genetic disorder characterized by congenital abnormalities, bone marrow failure and a heightened predisposition to cancer (19,20). It is a genetically heterogeneous disease, with 16 complementation groups identified to date (19,21,22). Our current understanding is that the products of these genes coordinately function to promote genome stability with a specialized role in the repair of DNA inter-strand crosslinks (ICLs; 20,23,24) and certain endogenous lesions (25). Activation of the FA pathway is typically observed by mono-ubiquitination of the FANCD2 and FANCI proteins by the FA core complex (composed of at least eight FA proteins), promoting their recruitment to chromatin and focus formation (26–29). Even in the absence of exogenous sources of ICLs, this activation occurs in normal S phase (29,30). Moreover, treatment of cells with a low dose of aphidicolin (APH), a polymerase inhibitor (31), robustly activates the FA pathway, indicating a role of the FA proteins during DNA replication (32). Previous studies reported that APH-induced FANCD2/FANCI foci often form as a pair (sister foci) during the G2/M phases, presumably flanking late replication intermediates at common fragile sites (33,34). These chromosomal loci are prone to breakage after partial inhibition of DNA replication (35), likely due to a paucity of DNA replication origins in these regions (36,37). As FA proteins are required for the stability of common fragile sites (32), they are likely to be involved in guiding successful replication of loci with fewer replication origins.
As Mcm4chaos3 homozygosity significantly decreases the total number of licensed origins on a genome-wide scale, it is likely to increase the number of loci lacking dormant origins or perhaps any origins. We therefore hypothesized that intrinsic activation of the FA pathway in Mcm4chaos3/chaos3 cells occurs in an attempt to support replication fork progression at these sites. To test this hypothesis, we introduced a null allele of Fancc (Fancc−), encoding a member of the FA core complex (38), into the Mcm4chaos3 background. Here, we report that loss of an intact FA pathway in Mcm4chaos3/chaos3 cells severely impairs replication fork stability even in unchallenged conditions. Furthermore, a reduced number of dormant origins also increased the number of sites in which replication was delayed until prophase, regardless of FA pathway activation. This increase in late replication intermediates as well as replication delay led to highly elevated levels of genome instability in Mcm4chaos3/chaos3;Fancc−/− cells and almost all Mcm4chaos3/chaos3;Fancc−/− pups died shortly after birth in an inbred C57BL/6J background. While viable in a mixed genetic background, Mcm4chaos3/chaos3;Fancc−/− mice still succumbed to spontaneous tumors at much younger ages than their Mcm4chaos3/chaos3 littermates. These findings confirm the role of dormant origins in stalled fork recovery while also unveiling their unique function in the complete replication of the genome, as other pathways are not able to fully compensate in their absence. Moreover, our results provide new insights into how the FA pathway functions in unchallenged conditions to promote genome stability.
MATERIALS AND METHODS
Mouse strains and MEFs
All experiments were performed using mice/cells derived from an inbred C57BL/6J genetic background or a C57BL/6J x C3HeB/FeJ-mixed genetic background and were approved by the Institutional Animal Care and Use Committee. Mouse embryonic fibroblasts (MEFs) were generated from 12.5–14.5 dpc embryos and cultured using standard procedures. All mice were genotyped by polymerase chain reaction. The primers used are available upon request.
Immunocytochemistry
Cells were grown on coverslips for 2 days and then fixed using 10% formalin. Whenever immunocytostaining was performed for replication protein A (RPA), cells were additionally pre-extracted with a 0.5% Triton X-100 in phosphate buffered saline solution for about 1 min prior to fixation. For analyses involving APH treatment, cells were given doses of either 150nM or 300nM APH for 24 h before harvest. Coverslips were then treated with the appropriate primary antibodies (4°C, overnight) and secondary antibodies (room temp, 1 h) and stained with 4',6-diamidino-2-phenylindole (DAPI, 1 μg/ml, 10 min) before being mounted onto slides using Vectashield (Vector Laboratories, #H-1000) or ProLong Gold antifade reagent (Life Technologies, #P36930). The Axio Imager A1 (Zeiss) was used for fluorescence microscopy analysis and to collect all images.
Antibodies
For immunocytochemistry, immunohistochemistry and western blotting procedures, we used anti-phospho-histone H3, anti-RPA32, anti-γH2AX (Cell Signaling; #9706, #2208 and #2577, respectively), anti-MCM4, anti-FANCD2, anti-FANCI, anti-53BP1 (Abcam; #ab4459, #ab2187, #ab74332 and #ab36823, respectively), anti-myeloperoxidase (Thermo Scientific; #RB-373-A1), anti-F4/80, anti-Mac-2 (Cedarlane; #CL8940AP and #CL8942AP, respectively), anti-cluster of differentiation 3 (Serotec; #MCA1477), anti-B220 (BD Biosciences; #550286), anti-digoxigenin (Roche; #11333062910), anti-FANCD2 (Epitomics; #2986-1), anti-PICH (Abnova; #H00054821-D01P) and Streptavidin-AlexaFluor488 conjugate antibody (Invitrogen, #S-32354). For the DNA fiber assay, the digoxigenin-rhodamine conjugate antibody from Roche (#11207750910) and Streptavidin-AlexaFluor488 conjugate antibody from Invitrogen (#S-32354) were used.
DNA fiber assay and cell cycle analysis
All techniques and methods of analysis used were performed as described previously (16,39).
EdU spots analysis
5-ethynyl-2'-deoxyuridine (EdU, Life Technologies, #A10044) was added to cells at a final concentration of 20μM for 10 min prior to harvest. After fixation, cells were subjected to the Click-iT™ reaction using Biotin-Azide (Life Technologies, #B10184) for 1 h at room temp, according to the manufacturer's instructions. Cells were then stained with the appropriate antibodies using normal immunocytochemistry techniques.
Cytokinesis-block micronucleus assay and 53BP1 nuclear bodies analysis
The cytokinesis-block micronucleus assays and 53BP1 nuclear bodies (53BP1-NBs) analyses were performed as described previously (40). Though the identification of G1 nuclei for the analysis of 53BP1-NBs is typically determined by those that are cyclin A-negative (41,42), the lack of a cyclin A antibody that works well for mouse cells precluded use of this technique. As an alternative, G1 nuclei were identified as those contained within binucleated cells following cytochalasin B treatment.
DNA ultra-fine bridge analysis
To measure the levels of DNA ultra-fine bridges (UFBs), another consequence of late replication intermediates (33,34,43), anaphase cells were stained for the Pkl1-interacting checkpoint helicase (PICH) and analyzed for the presence of PICH-coated UFBs.
RESULTS
Mcm4chaos3 homozygosity results in a reduced number of dormant origins and a lower active origin density in a C57BL/6J background
The phenotypes of Mcm4chaos3/chaos3 and Fancc−/− mice in a C57BL/6J background have been relatively well characterized and share several similarities, including semi-lethality and a heightened predisposition to microphthalmia (39,40,44). We therefore generated wild-type, Mcm4chaos3/chaos3, Fancc−/− and Mcm4chaos3/chaos3;Fancc−/− primary MEFs in this inbred background to perform various types of cellular assays. First, we verified that Mcm4chaos3/chaos3 and Mcm4chaos3/chaos3;Fancc−/− cells have reduced levels of chromatin-bound MCM4 compared to wild-type and Fancc−/− cells, indicating a reduced number of licensed origins (Supplementary Figure S1A) (16). Consistent with our previous work (16), Mcm4chaos3/chaos3 cells exhibited higher levels of FANCD2 foci at prophase compared to wild-type cells (Supplementary Figure S1B). Expectedly, the lack of FANCC abolished FANCD2 chromatin loading to sub-detectable levels in both Fancc−/− and Mcm4chaos3/chaos3;Fancc−/− cells (Supplementary Figure S1C). To understand DNA replication kinetics in these cells, we began our analysis with a DNA fiber assay. Previously, we reported that Mcm4chaos3 homozygosity in this inbred background not only results in a reduced number of dormant origins but also lowers the density of active origins, thereby contributing to the semi-lethality of newborn mice (39). To verify that this was also the case for Mcm4chaos3/chaos3;Fancc−/− cells, we measured origin-to-origin distances (Figure 1A and B). Compared to wild-type cells (63.3 kb ± 3.57), significantly longer average origin-to-origin distances were observed in Mcm4chaos3/chaos3 (80.0 kb ± 4.32, P < 0.01) and Mcm4chaos3/chaos3;Fancc−/− cells (75.0 kb ± 3.60, P < 0.05), indicating lower densities of active origins. While Fancc−/− cells exhibited a slightly longer average origin-to-origin distance (68.3 kb ± 4.12), this was not statistically different from wild-type cells (P = 0.361). To determine if overall replication fork movement was substantially altered in mutant cells, fork velocities were also measured (Figure 1A). Average fork velocities were largely similar among the four genotypes, with only Fancc−/− and Mcm4chaos3/chaos3;Fancc−/− cells exhibiting very slightly faster and slower forks, respectively (Figure 1B).
Figure 1.

(A) A schematic of the consecutive dual labeling steps of the DNA fiber assay is shown at left. Active replication forks were labeled with deoxyuridine triphophates (dUTPs) conjugated with digoxigenin (250μM; red) for 20 min followed by labeling with biotin-conjugated dUTPs (250μM; green) for 30 min. Fork velocity and origin-to-origin distances were determined as shown. A representative image of adjacent origins is shown on the right with staining for digoxigenin-dUTPs (red) and biotin-dUTPs (green). The white arrowheads indicate the location of origins. The scale bar is 10 μm. (B) Box plots show the ranges observed for the origin-to-origin (ori-to-ori) distance (left) and fork velocity (right) values for each genotype. Lines within the shaded boxes indicate the medians while the ‘+’ signs show the location of the mean. Black dots represent outliers. The tables on the right show the summaries for ori-to-ori distances (top) and fork velocities (bottom). Significance was determined by t-test. The asterisks denote: *P < 0.05, **P < 0.01 and ***P < 0.001. NS means not significant. WT, C3, FAC and C3;FAC refer to wild-type, Mcm4chaos3/chaos3, Fancc−/− and Mcm4chaos3/chaos3;Fancc−/−, respectively.
Loss of FANCC leads to a drastic increase in stalled/collapsed forks in Mcm4chaos3/chaos3 cells
The FA proteins function to support replication fork stability as well as fork restart (30,45–47). Thus, Mcm4chaos3/chaos3;Fancc−/− cells lack two important fork recovery mechanisms: dormant origins and another mechanism mediated by FANCC. To investigate fork progression under these circumstances, we examined the focus formation of RPA32 (Figure 2A), a marker of stalled replication forks (48,49). Compared to wild-type cells, slightly increased numbers of Mcm4chaos3/chaos3 and Fancc−/− cells were positive for ≥5 RPA32 foci (1.2- and 1.6-fold, respectively) though only Fancc−/− cells exhibited a significant increase (P < 0.001), similar to our previous observations (40). The marginal increase in RPA32 foci in Mcm4chaos3/chaos3 cells may be due to intrinsic upregulation of the FA pathway, which could compensate for a loss of dormant origins in the recovery of stalled/collapsed forks. Indeed, loss of FANCC in Mcm4chaos3/chaos3 cells resulted in drastically increased levels of RPA32 foci (>3-fold, P < 0.001, relative to wild-type cells). Furthermore, the number of RPA32 foci co-localizing with γH2AX foci, a marker of DNA double strand breaks (50), exhibited a similar trend with the highest increase (>3.8-fold, P < 0.001, relative to wild-type cells) seen in Mcm4chaos3/chaos3;Fancc−/− cells. These findings suggest that FANCC plays a crucial role in preventing stalled/collapsed forks, particularly in the absence of dormant origins. We then investigated the ability of Mcm4chaos3/chaos3;Fancc−/− cells to progress through the cell cycle. Cell cycle analysis reproduced a typical profile of Mcm4chaos3/chaos3 cells (17,39,51) including a reduced S phase fraction (24.6% versus 28.1% in wild-type, P < 0.01) and G2/M accumulation (24.1% versus 21.1% in wild-type, P < 0.01) (Figure 2B; Supplementary Figure S2). While cell cycle profiles for Fancc−/− cells were indistinguishable from Mcm4chaos3/chaos3 cells, these trends were further exacerbated in Mcm4chaos3/chaos3;Fancc−/− cells, in which the S phase and G2/M fractions were 19.3% and 28.0%, respectively (P < 0.001). Mcm4chaos3/chaos3;Fancc−/− cells therefore suffer from a greatly increased level of late replication intermediates, accumulating during the G2/M phases.
Figure 2.

(A) Mcm4chaos3/chaos3;Fancc−/− cells display a drastic increase in markers of persistently stalled replication forks upon exiting S phase. Shown at left are representative images of all four genotypes co-stained for RPA (green) and γH2AX (red). The average percentages of cells positive for five or more RPA foci (top right) or five or more RPA–γH2AX co-localizations (bottom right) are also shown. Error bars show the binomial error for the combined data set obtained from three independently performed experiments. The scale bar is 10 μm. Significance was determined by χ2-test. (B) Mcm4chaos3/chaos3;Fancc−/− cells experience a heightened accumulation in the G2/M phases. Shown are the average proportions of cells observed within the G1, S or G2/M phases for all four genotypes. Error bars show the standard error of the mean (SEM) for at least five independent replicates. The asterisks denote: *P < 0.05, **P < 0.01 and ***P < 0.001. WT, C3, FAC and C3;FAC refer to wild-type, Mcm4chaos3/chaos3, Fancc−/− and Mcm4chaos3/chaos3;Fancc−/−, respectively.
Despite FA pathway activation, Mcm4chaos3/chaos3 cells exhibit an increase in sites of isolated DNA synthesis in early M phase
Although Mcm4chaos3/chaos3 cells exhibited only a marginal increase in RPA32/γH2AX foci, they still exhibited significant G2/M phase accumulation (Figure 2B). This may therefore stem from an additional defect, such as delayed completion of DNA replication. Recent studies reported that delayed DNA replication can be identified as sites of localized DNA synthesis persisting until early M phase by pulse-labeling with EdU, a thymidine analog (referred to as ‘EdU spots’; 52,53). We therefore quantified EdU spots in prophase cells after a short pulse labeling (10 min) of EdU. We also examined the co-localization of EdU spots with FANCD2 foci in wild-type and Mcm4chaos3/chaos3 cells (Figure 3A), as EdU spots co-localize with FANCD2 foci (52,53) and FANCI foci (Supplementary Figure S3A) in early M phase. Compared to wild-type cells, a greatly elevated percentage (>3-fold, P < 0.001) of Mcm4chaos3/chaos3 cells contained prophase EdU spots even in the untreated condition (Figure 3B). Consistent with previous studies (52,53), the majority of EdU spots indeed co-localized with FANCD2 foci in either genotype, resulting in a corresponding increase in the number of Mcm4chaos3/chaos3 cells positive for EdU-FANCD2 co-localizations. This was further supported by the analysis of individual EdU spots, which showed that only a minor fraction failed to co-localize with FANCD2 in either genotype (Figure 3C), possibly representing loci that are naturally late replicating. Interestingly, analysis of individual FANCD2 foci revealed an almost exclusive increase in those co-localizing with EdU spots in Mcm4chaos3/chaos3 cells (Figure 3C). Consistent with previous findings (52), we also observed that treatment with a low dose of APH greatly elevated the numbers of both EdU spots and FANCD2 foci in both genotypes (Supplementary Figure S3B), yielding trends very similar to the untreated condition. Taken together, these observations indicate that the elevated number of FANCD2 foci seen in Mcm4chaos3/chaos3 cells is primarily due to an increased number of sites exhibiting isolated DNA synthesis, though this activation is apparently insufficient to prevent this synthesis from being delayed until prophase. We interpret these findings as follows: a loss of dormant origins not only lowers the efficiency of stalled fork recovery but also generates long, origin-poor regions. The FA pathway can effectively compensate for the former, thus preventing the formation of RPA32/γH2AX foci as seen in Mcm4chaos3/chaos3 cells (Figure 2A). In the latter case, however, it is almost inevitable that fork stalling occurs and persists more frequently in origin-poor regions, thereby delaying the completion of DNA replication in these regions until early M phase despite FA pathway activation.
Figure 3.

A higher number of prophase FANCD2 foci in Mcm4chaos3/chaos3 cells are the result of a sharp increase in EdU spots that co-localize with FANCD2. (A) Shown are representative images of wild-type and Mcm4chaos3/chaos3 cells co-stained for EdU (green) and FANCD2 (red) in the untreated (UNT) condition. Prophase cells were identified by prominent chromatin condensation via DAPI staining (blue). The scale bar is 10 μm. (B) At top are the percentages of prophase cells positive for EdU spots (white bars) or EdU-FANCD2 co-localization events (gray bars). At bottom are the number of EdU spots (white bars) per prophase cell and the number of EdU-FANCD2 co-localization events (gray bars) per prophase cell. Error bars show the binomial error for the combined data sets obtained from three independently performed experiments. Significance was determined by χ2-test. The asterisks denote: ***P < 0.001. (C) Shown at top are the numbers of EdU spots with ‘EdU only’ (white bars) and the number of EdU-FANCD2 co-localization events (gray bars) observed in 150 prophase cells. At bottom are the numbers of FANCD2 foci with ‘FANCD2 only’ (black bars) and the number of EdU-FANCD2 co-localizations (shown again as gray bars) observed in 150 prophase cells. WT and C3 refer to wild-type and Mcm4chaos3/chaos3, respectively.
The FA pathway exerts its role in preventing delayed DNA replication under conditions of replication stress
Based on our interpretations, activation of the FA pathway should act against the formation of EdU spots at prophase, though its effect may be limited. On the other hand, given the high level of EdU-FANCD2 co-localizations, an alternative possibility is that FANCD2 mono-ubiquitination is actually required for this isolated form of DNA synthesis. To distinguish between these two possibilities, we repeated our pulse-labeling experiments to score prophase EdU spots in all four genotypes in the untreated conditions (Figure 4A and B). A reproducibly elevated percentage (>2-fold, P < 0.001) of Mcm4chaos3/chaos3 cells was positive for EdU spots relative to wild-type cells, whereas Fancc−/− cells displayed no significant difference from wild-type (P = 0.251), consistent with the idea that mono-ubiquitinated FANCD2 is dispensable for this isolated DNA synthesis. Interestingly, however, a slightly increased percentage of Mcm4chaos3/chaos3;Fancc−/− cells contained EdU spots compared to Mcm4chaos3/chaos3 cells (P < 0.01), suggesting that FANCC is actually required to prevent the formation of EdU spots in the absence of dormant origins. To further test the role of the FA pathway in preventing this delayed replication, we treated cells with a low dose of APH, which substantially increased the number of EdU spots in all genotypes (Figure 4C and D). A significantly increased number (1.5-fold compared to wild-type cells, P < 0.001) of Fancc−/− cells were now positive for ≥10 APH-induced EdU spots. As APH is known to slow fork velocities (36,53), increase the frequency of fork stalling (16,54) and hyperactivate dormant origins (14,16), it seems likely that such conditions make the role of the FA pathway more crucial to sustain fork progression and prevent delayed replication. Indeed, we observed a sharp increase in the number of Mcm4chaos3/chaos3;Fancc−/− cells exhibiting ≥10 APH-induced EdU spots (1.9-fold increase compared to wild-type cells, P < 0.001), with a 2-fold higher number of APH-induced EdU spots per cell compared to all other genotypes. These data support the idea that FANCC, and potentially other components of the FA pathway, functions to prevent delayed DNA replication, particularly under conditions of replication stress.
Figure 4.
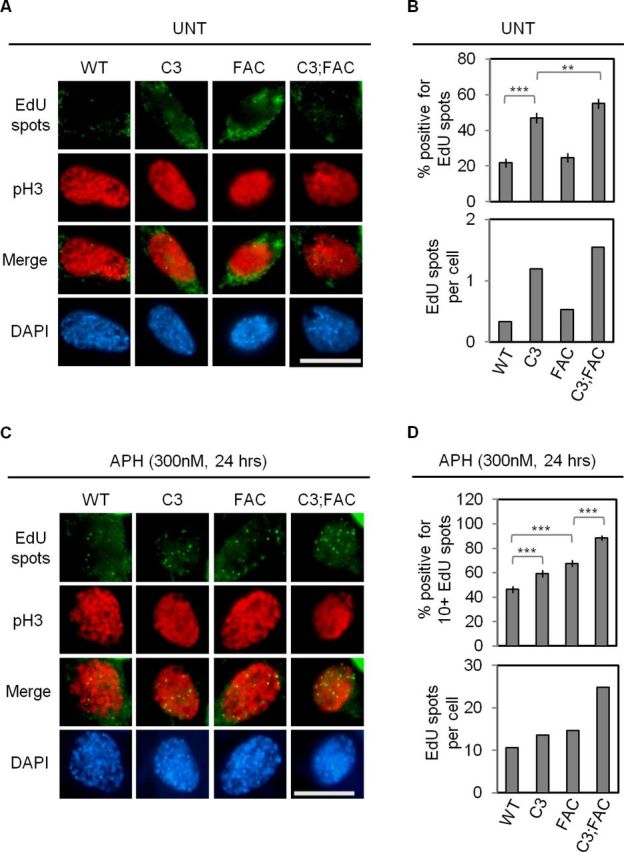
A loss of dormant origins increases the number of EdU spots at prophase, which is further enhanced by disruption of Fancc. (A) Shown are representative images of all four genotypes co-stained for EdU (green) and pH3 (red). In this experiment, staining for pH3, a marker of condensed chromatin (67), was used to confirm the scoring of nuclei at prophase in addition to nuclear morphology by DAPI staining. (B) Shown are the average percentages of prophase cells positive for EdU spots (top) and the number of EdU spots per prophase cell (bottom). (C) As in (A), with cells treated with 300nM APH for 24 h. (D) Shown are the average percentages of prophase cells positive for 10 or more EdU spots (top) and the number of EdU spots per prophase cell (bottom). Nuclei (A,C) were stained with DAPI (blue). Scale bars (A,C) are 10 μm. Error bars (B,D) show the binomial error for the combined data sets obtained from three independently performed experiments. Significance was determined by χ2-test. The asterisks denote: **P < 0.01 and ***P < 0.001. WT, C3, FAC and C3;FAC refer to wild-type, Mcm4chaos3/chaos3, Fancc−/− and Mcm4chaos3/chaos3;Fancc−/−, respectively.
The frequencies of spontaneous MN and 53BP1-NBs are significantly increased in Mcm4chaos3/chaos3;Fancc−/− cells
Based on the above findings, we conclude that Mcm4chaos3/chaos3;Fancc−/− cells suffer from two distinct defects: (i) a greatly increased level of stalled/collapsed forks due to the lack of two major fork recovery mechanisms (dormant origins and the FA pathway) and (ii) an increased number of sites in which replication is delayed until early M phase. To investigate the consequences of these accumulated late replication intermediates, we first looked at the formation of MN, which occur at an elevated frequency in Mcm4chaos3/chaos3 and Fancc−/− cells (16,40). Using the cytokinesis-block micronucleus assay (55), we observed that Mcm4chaos3/chaos3 cells reproducibly exhibited a 2.9-fold increase in spontaneous MN compared to wild-type cells (P < 0.001; Figure 5A). Fancc−/− cells showed an even higher increase (3.5-fold, P < 0.001), with the most severe phenotype seen for Mcm4chaos3/chaos3;Fancc−/− cells (5.3-fold, P < 0.001). If unresolved, late replication intermediates can also manifest as 53BP1-NBs in the subsequent G1 phase nuclei (41,42). These structures co-localize with γH2AX and are often exquisitely symmetrical in terms of their appearance within the daughter nuclei, suggesting that they are derived from a common breakage event occurring during passage through M phase (41). We thus measured the levels of 53BP1-NBs in G1 phase daughter nuclei contained within binucleated cells using essentially the same protocol as the cytokinesis-block micronucleus assay. While Mcm4chaos3/chaos3 and Fancc−/− cells displayed slightly increased numbers (1.16- and 1.5-fold, respectively) of G1 nuclei positive for 53BP1-NBs (P < 0.01 and P < 0.001, respectively) compared to wild-type, Mcm4chaos3/chaos3;Fancc−/− cells had a substantial increase in 53BP1-NBs compared to either single mutant (1.9-fold, P < 0.001) (Figure 5B). Taken together, these findings suggest that FANCC and dormant origins coordinately function to prevent genome instability derived from late replication intermediates.
Figure 5.

Mcm4chaos3/chaos3;Fancc−/− cells show highly elevated levels of MN and 53BP1-NBs. (A) Shown are a representative image of binucleated cell positive for a micronucleus (left), the average percentages of binucleated cells positive for MN (middle) and a distribution of the number of MNs per binucleated cell (right). (B) Shown at left are representative images of a 53BP1-NB in a binucleated cell. 53BP1 staining is in red. At middle are the average percentages of individual nuclei from binucleated cells positive for 53BP1-NBs, and at right are the distributions indicating the number of 53BP1-NBs per G1 phase nucleus. All nuclei were stained with DAPI (blue). All scale bars are 10 μm. All error bars show the binomial error for the combined data sets obtained from three independently performed experiments. Significance was determined by χ2-test. The asterisks denote: *P < 0.05, **P < 0.01 and ***P < 0.001. WT, C3, FAC and C3;FAC refer to wild-type, Mcm4chaos3/chaos3, Fancc−/− and Mcm4chaos3/chaos3;Fancc−/−, respectively.
Almost all B6 Mcm4chaos3/chaos3;Fancc−/− pups die right after birth
Given the additive effect of Fancc− and Mcm4chaos3 in causing genome instability, we hypothesized that B6 Mcm4chaos3/chaos3;Fancc−/− mice may exhibit more severe phenotypes than either single homozygote. We therefore set up Fancc+/− heterozygous intercrosses in the Mcm4chaos3 homozygous background to obtain B6 double homozygous mutants. Among 74 newborns, the numbers of pups for the three expected genotypes were not statistically different from the expected Mendelian ratio (P = 0.640; see Table 1). However, 13 out of 15 double homozygous (Mcm4chaos3/chaos3;Fancc−/−) pups died shortly after birth, revealing a strikingly high rate of perinatal lethality. We previously reported that Mcm4chaos3 and Fancc− homozygosity each cause semi-lethality in this background (39,40). However, this cannot fully explain the high lethality of Mcm4chaos3/chaos3;Fancc−/− pups, as their survival rate (13%) was substantially lower than that of their Mcm4chaos3/chaos3;Fancc+/+ littermates (85%, P = 7.64 ×10−15) or of Fancc−/− mice (65%) (40). We also noticed that Mcm4chaos3/chaos3;Fancc+/− pups exhibited a decreased rate of survival (69%) at 3 weeks of age compared to Mcm4chaos3/chaos3;Fancc+/+ pups (P = 0.00582). This Fancc dosage-dependent survival of Mcm4chaos3/chaos3 pups supports a synthetic lethal/sickness interaction between Mcm4chaos3 and Fancc−. Histopathological analyses of Mcm4chaos3/chaos3;Fancc−/− pups did not reveal an apparent cause of death. Of the two Mcm4chaos3/chaos3;Fancc−/− pups that survived past 3 weeks, one was euthanized for a non-tumor abscess at the age of 128 days while the other developed disseminated T-cell lymphoma in only 163 days (Supplementary Figure S4), much more quickly than the average tumor latency of about 12.4 months for Mcm4chaos3/chaos3 mice in the B6 background (16). However, due to the very high rate of perinatal lethality, it has been extremely difficult to generate additional Mcm4chaos3/chaos3;Fancc−/− pups even after the expansion of mating crosses. Together, these data indicate that a concomitant loss of dormant origins and Fancc is incompatible with postnatal development in this inbred background.
Table 1. Mcm4chaos3/chaos3;Fancc−/− mice exhibit severe perinatal lethality in an inbred C57BL/6J background.
| Genotype (all in Mcm4chaos3/chaos3) | Number of pups found at birth | Number of live pups | Survival at 3 weeks | P value | |
|---|---|---|---|---|---|
| At birth | At 3 weeks | ||||
| Fancc +/+ | 20 | 18 | 17 | 85% | – |
| Fancc +/− | 39 | 31 | 27 | 69% | 0.00582 |
| Fancc −/− | 15 | 2 | 2 | 13% | 7.64E-15 |
| Total | 74 | 51 | 46 | 62% | |
Mcm4chaos3/chaos3;Fancc−/− mice are viable in a mixed genetic background but succumb to spontaneous tumors at much younger ages
It is well known that phenotypic expression in mice is greatly influenced by genetic background. We therefore reasoned that performing the same intercrosses in a different genetic background would allow us to obtain viable Mcm4chaos3/chaos3;Fancc−/− mice to test their tumor predisposition. Indeed, we found Mcm4chaos3/chaos3;Fancc−/− mice were viable when generated in a mixed background between C57BL/6J and C3HeB/FeJ. Fancc−/− and Mcm4chaos3/chaos3;Fancc−/− mice were again born at reduced frequencies compared to the expected numbers (Supplementary Tables S1 and S2), but these differences were not statistically significant. While Mcm4chaos3/chaos3;Fancc−/− mice did not show any apparent developmental abnormalities, they did exhibit a slightly decreased average body weight (P < 0.05; Figure 6A). A loss of dormant origins also worsened the hypogonadism phenotype of Fancc−/− mice, as Mcm4chaos3/chaos3;Fancc−/− mice exhibited a smaller average testes size (P < 0.05 when compared to Fancc−/− mice) with an increased number of empty seminiferous tubules (Supplementary Figure S5A and B). After verifying an enhanced level of replication-associated genome instability in Mcm4chaos3/chaos3;Fancc−/− MEFs in this background (Supplementary Figure S6), we aged cohorts of wild-type, Mcm4chaos3/chaos3, Fancc−/− and Mcm4chaos3/chaos3;Fancc−/− mice to observe the formation of spontaneous tumors until 14 months of age. In agreement with previous findings (44,56), >95% of wild-type and Fancc−/− mice survived to the end of the study, with only one Fancc−/− mouse exhibiting tumors at necropsy (Figure 6B). As expected, the majority (∼71%) of Mcm4chaos3/chaos3 mice succumbed to tumors before 14 months, as did ∼84% of Mcm4chaos3/chaos3;Fancc−/− mice. The tumor spectrum displayed by Mcm4chaos3/chaos3;Fancc−/− mice was largely similar to that of Mcm4chaos3/chaos3 mice, though the entire cohort that developed tumors before 235 days succumbed exclusively to lymphosarcomas (Figure 6C and Supplementary Tables S3 and S4). Most of these tumors were of T-cell origin (CD-3 positive), though B-cell tumors (B220 positive) were also observed (Supplementary Figure S7). Notably, a myeloid leukemia was seen as well. Overall, Mcm4chaos3/chaos3;Fancc−/− mice exhibited an average tumor latency of 279.7 days, much shorter than the 361.9 days seen for Mcm4chaos3/chaos3 mice. This difference did not reach statistical significance (log-rank test, P = 0.264), most likely due to great variation in the latencies in individual mice and gender-specific effects in this background (Supplementary Tables S3 and S4) (57). However, it should be noted that a much greater proportion of Mcm4chaos3/chaos3;Fancc−/− mice (∼53%) developed spontaneous tumors before 300 days of age compared to Mcm4chaos3/chaos3 mice (∼7%; P = 1.21 × 10−19) suggesting that a concomitant loss of dormant origins and Fancc can accelerate tumorigenesis.
Figure 6.

(A) The average body weight of Mcm4chaos3/chaos3;Fancc−/− mice derived in a C57BL/6J × C3HeB/FeJ mixed genetic background is slightly reduced. Shown are the average body weights of the four genotypes in grams. Error bars show the standard error of the means (SEMs) for at least five mice per genotype. Significance was determined by t-test. The asterisks denote: *P < 0.05. (B) Mcm4chaos3/chaos3;Fancc−/− mice succumb to tumors with a shorter latency than Mcm4chaos3/chaos3 mice in a C57BL/6J x C3HeB/FeJ-mixed genetic background. Shown are the tumor-free survival curves for all four genotypes. (C) Shown are the tumor spectra observed for Mcm4chaos3/chaos3 and Fancc−/−;Mcm4chaos3/chaos3 mice. WT, C3, FAC and C3;FAC refer to wild-type, Mcm4chaos3/chaos3, Fancc−/− and Mcm4chaos3/chaos3;Fancc−/−, respectively.
DISCUSSION
Exploiting the Mcm4chaos3 mouse model, we investigated the nature of FA pathway activation in the absence of dormant origins. We hypothesized that this activation occurs to compensate for the rescue of stalled forks. In agreement with our hypothesis, Mcm4chaos3/chaos3;Fancc−/− cells exhibited impaired fork stability, an increased number of sites displaying delayed replication and greatly enhanced levels of genome instability. Furthermore, we found that an intact FA pathway is required in Mcm4chaos3/chaos3 mice not only for promoting tumor suppression but also for supporting postnatal development under conditions in which the density of active origins is relatively low (39).
While the exact role of the FA pathway in unchallenged S phase is still largely unknown, a loss of Fancc in Mcm4chaos3/chaos3 cells clearly revealed its function in replication fork progression as well as stability. In the absence of dormant origins, the FA core complex may facilitate translesion synthesis to recover stalled forks (58–60). Another recent study demonstrated a role of FANCD2 in BLM-mediated fork restart (47). This mechanism may come into play in the absence of dormant origins. Alternatively, if no appropriate choice is available for stalled fork recovery during S phase, the FA pathway may function to stabilize stalled forks, thereby manifesting as FANCD2/FANCI foci during the G2/M phases, as proposed earlier (33). Indeed, lack of a functional FA pathway leads to the degradation of nascent strands at stalled forks, supporting its role in fork protection (46,47,61). Furthermore, given the increased levels of spontaneous genome instability in Fancc−/− cells, it seems that there must be certain types of endogenous lesions that are primarily taken care of by the FA pathway rather than dormant origins. Fork recovery at such lesions may also be shared with HELQ, as double homozygosity for Fancc− and Helqgt greatly elevates genome instability (40).
In this study, we observed that an elevated level of FANCD2 focus formation in Mcm4chaos3/chaos3 cells during prophase (Supplementary Figure S1B) is primarily associated with the formation of EdU spots (Figure 3). A recent study reported that polymerase eta deficiency induces common fragile site instability and the formation of EdU spots co-localizing with FANCD2 foci in early M phase, suggesting that EdU spots most likely represent delayed DNA replication within loci where replication intermediates long persist (52). Consistent with this idea, APH treatment predominantly induced EdU spots co-localizing with FANCD2 foci both in wild-type and Mcm4chaos3/chaos3 cells (Figure 3). Despite the high rate of EdU-FANCD2 co-localizations, an intact FA pathway is apparently dispensable for this type of DNA synthesis. On the contrary, lack of a functional FA pathway actually increases the number of prophase EdU spots under conditions of replication stress, as seen in Mcm4chaos3/chaos3;Fancc−/− cells in the untreated condition and both Fancc−/− and Mcm4chaos3/chaos3;Fancc−/− cells in the APH-treated condition (Figure 4). These findings suggest the presence of an FA-independent mechanism(s) to support DNA synthesis at such loci until early M phase. Such mechanisms may include HELQ, which functions parallel to FANCC, as we reported very recently (40). Moreover, it was recently shown that the structure-specific endonucleases MUS81 and ERCC1 are also found at EdU spots along with FANCD2 but their presence on mitotic chromosomes does not depend on FANCC (53). While the exact mechanism(s) remains to be unveiled, it was demonstrated that depletion of these endonucleases increased the formation of MN, DNA UFBs at anaphase and 53BP1-NBs (53,62). It will therefore be important to understand the roles of these endonucleases in the formation of EdU spots as well as the role of this late DNA synthesis in the resolution of late replication intermediates.
Mcm4chaos3/chaos3 and Fancc−/− cells/mice are phenotypically similar, particularly in the B6 background, with respect to genome instability (Figure 5) and susceptibility to newborn lethality and microphthalmia (39,44). However, Mcm4chaos3/chaos3 mice are highly prone to spontaneous tumorigenesis while Fancc−/− mice are not. One major phenotype that was found in Mcm4chaos3/chaos3 but not Fancc−/− cells was an increased number of EdU spots in prophase in the untreated condition (Figure 4B). We thus speculate that a reduction in the number of licensed origins by Mcm4chaos3 homozygosity not only causes a loss of dormant origins but also generates relatively long stretches of the genome that are devoid of any replication origins. So, fork stalling within small regions lacking dormant origins may be fully replicated by the fork rescuing actions of the FA pathway described above. However, this activity may not be sufficient in large, origin-poor loci so that completion of DNA synthesis in these regions is delayed until early M phase despite FA pathway activation, thus manifesting as a large increase in the number of EdU-FANCD2 co-localizations. It is possible that a fraction of these sites may not even fully complete DNA replication prior to anaphase. Therefore, a combination of intrinsic chromosome instability along with an increased number of un-replicated loci could be a driving factor in spontaneous tumorigenesis in Mcm4chaos3/chaos3 mice.
While several mouse models have been generated to study the FA pathway, the majority fails to recapitulate the phenotypes of human FA patients, including tumor predisposition (63,64). We were unable to further test to what extent Mcm4chaos3/chaos3;Fancc−/− mice recapitulate other phenotypes of human FA patients, such as bone marrow failure. However, we think that Mcm4chaos3 homozygosity provides a unique condition to understand the role of the FA proteins as well as others in genome stability in unchallenged conditions, as recently shown for ATM (57). In particular, an FA core complex-independent role of FANCD2 in replication fork stability (61) can also be investigated in Mcm4chaos3/chaos3 mice, which may clarify the role of FANCD2 in the formation of EdU spots. Very recently, the first human genetic disorder caused by a mutant MCM4 gene was discovered (65,66). Cells from these patients exhibit chromosome fragility much like Mcm4chaos3/chaos3 cells. Due to a very limited number of patients with this disorder, however, it is not yet unknown whether they are cancer-prone. Nevertheless, it is quite possible that there remain undiscovered genetic disorders caused by mutations in MCM2-7 genes in which the physiological role of the FA pathway may become more apparent.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgements
We thank Dr Markus Grompe for providing us with the Fancc− mice, Dr Alexandra Sobeck for her critical reading of this manuscript and Katie Haberle, Masanao Chinen, Susan Yeung and Wai Long Lee for their assistance in genotyping.
Conflict of interest statement. The authors declare that they have no conflict of interest.
Footnotes
These authors contributed equally to this work.
FUNDING
National Institutes of Health [R01CA148806 to N.S.]; Japan Society for the Promotion of Science [to T.K.].
REFERENCES
- 1.Yekezare M., Gómez-González B., Diffley J.F.X. Controlling DNA replication origins in response to DNA damage—inhibit globally, activate locally. J. Cell Sci. 2013;126:1297–1306. doi: 10.1242/jcs.096701. [DOI] [PubMed] [Google Scholar]
- 2.Blow J.J., Dutta A. Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell Biol. 2005;6:476–486. doi: 10.1038/nrm1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sclafani R.A., Holzen T.M. Cell cycle regulation of DNA replication. Annu. Rev. Genet. 2007;41:237–280. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Labib K., Tercero J.A., Diffley J.F.X. Uninterrupted MCM2-7 function required for DNA replication fork progression. Science. 2000;288:1643–1647. doi: 10.1126/science.288.5471.1643. [DOI] [PubMed] [Google Scholar]
- 5.Pacek M., Walter J.C. A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J. 2004;23:3667–3676. doi: 10.1038/sj.emboj.7600369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shechter D., Ying C.Y., Gautier J. DNA unwinding is an MCM complex-dependent and ATP hydrolysis-dependent process. J. Biol. Chem. 2004;279:45586–45593. doi: 10.1074/jbc.M407772200. [DOI] [PubMed] [Google Scholar]
- 7.Moyer S.E., Lewis P.W., Botchan M.R. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. U.S.A. 2006;103:10236–10241. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ilves I., Petojevic T., Pesavento J.J., Botchan M.R. Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol. Cell. 2010;37:247–258. doi: 10.1016/j.molcel.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burkhart R., Schulte D., Hu B., Musahl C., Göhring F., Knippers R. Interactions of human nuclear proteins P1Mcm3 and P1Cdc46. Eur. J. Biochem. 1995;228:431–438. [PubMed] [Google Scholar]
- 10.Rowles A., Chong J.P.J., Brown L., Howell M., Evan G.I., Blow J.J. Interaction between the origin recognition complex and the replication licensing system in Xenopus. Cell. 1996;87:287–296. doi: 10.1016/s0092-8674(00)81346-x. [DOI] [PubMed] [Google Scholar]
- 11.Mahbubani H.M., Chong J.P.J., Chevalier S., Thömmes P., Blow J.J. Cell cycle regulation of the replication licensing system: involvement of a Cdk-dependent inhibitor. J. Cell Biol. 1997;136:125–135. doi: 10.1083/jcb.136.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards M.C., Tutter A.V., Cvetic C., Gilbert C.H., Prokhorova T.A., Walter J.C. MCM2-7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J. Biol. Chem. 2002;277:33049–33057. doi: 10.1074/jbc.M204438200. [DOI] [PubMed] [Google Scholar]
- 13.Woodward A.M., Göhler T., Luciani M.G., Oehlmann M., Ge X., Gartner A., Jackson D.A., Blow J.J. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell. Biol. 2006;173:673–683. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge X.Q., Jackson D.A., Blow J.J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007;21:3331–3341. doi: 10.1101/gad.457807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibarra A., Schwob E., Méndez J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8956–8961. doi: 10.1073/pnas.0803978105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawabata T., Luebben S.W., Yamaguchi S., Ilves I., Matise I., Buske T., Botchan M.R., Shima N. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell. 2011;41:543–553. doi: 10.1016/j.molcel.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shima N., Alcaraz A., Liachko I., Buske T.R., Andrews C.A., Munroe R.J., Hartford S.A., Tye B.K., Schimenti J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007;39:93–98. doi: 10.1038/ng1936. [DOI] [PubMed] [Google Scholar]
- 18.Blow J.J., Ge X.Q., Jackson D.A. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011;36:405–414. doi: 10.1016/j.tibs.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crossan G.P., Patel K.J. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J. Pathol. 2012;226:326–337. doi: 10.1002/path.3002. [DOI] [PubMed] [Google Scholar]
- 20.Kottemann M.C., Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kashiyama K., Nakazawa Y., Pilz D.T., Guo C., Shimada M., Sasaki K., Fawcett H., Wing J.F., Lewin S.O., Carr L., et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 2013;92:807–819. doi: 10.1016/j.ajhg.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bogliolo M., Schuster B., Stoepker C., Derkunt B., Su Y., Raams A., Trujillo J.P., Minguillón J., Ramírez M.J., Pujol R., et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013;92:800–806. doi: 10.1016/j.ajhg.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Constantinou A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma. 2012;121:21–36. doi: 10.1007/s00412-011-0349-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kee Y., D’Andrea A.D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–1694. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langevin F., Crossan G.P., Rosado I.V., Arends M.J., Patel K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–58. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Higuera I., Taniguchi T., Ganesan S., Meyn M.S., Timmers C., Hejna J., Grompe M., D’Andrea A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 27.Sims A.E., Spiteri E., Sims R.J., Arita A.G., Lach F.P., Landers T., Wurm M., Freund M., Neveling K., Hanenberg H., et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2007;14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- 28.Smogorzewska A., Matsuoka S., Vinciguerra P., McDonald E.R., III, Hurov K.E., Luo J., Ballif B.A., Gygi S.P., Hofmann K., D’Andrea A.D., et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taniguchi T., Garcia-Higuera I., Andreassen P.R., Gregory R.C., Grompe M., D’Andrea A.D. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002;100:2414–2420. doi: 10.1182/blood-2002-01-0278. [DOI] [PubMed] [Google Scholar]
- 30.Sobeck A., Stone S., Costanzo V., de Graaf B., Reuter T., de Winter J., Wallisch M., Akkari Y., Olson S., Wang W., et al. Fanconi anemia proteins are required to prevent accumulation of replication-associated DNA double-strand breaks. Mol. Cell. Biol. 2006;26:425–437. doi: 10.1128/MCB.26.2.425-437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikegami S., Taguchi T., Ohashi M., Nagano H., Mano Y. Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-alpha. Nature. 1978;275:458–460. doi: 10.1038/275458a0. [DOI] [PubMed] [Google Scholar]
- 32.Howlett N.G., Taniguchi T., Durkin S.G., D’Andrea A.D., Glover T.W. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- 33.Chan K.L., Palmai-Pallag T., Ying S., Hickson I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell. Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 34.Naim V., Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell. Biol. 2009;11:761–768. doi: 10.1038/ncb1883. [DOI] [PubMed] [Google Scholar]
- 35.Durkin S.G., Glover T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 36.Letessier A., Millot G.A., Koundrioukoff S., Lachages A.-M., Vogt N., Hansen R.S., Malfoy B., Brison O., Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011;470:120–123. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- 37.Ozeri-Galai E., Lebofsky R., Rahat A., Bester A.C., Bensimon A., Kerem B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell. 2011;43:122–131. doi: 10.1016/j.molcel.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 38.Whitney M., Royle G., Low M., Kelly M., Axthelm M., Reifsteck C., Olson S., Braun R., Heinrich M., Rathbun R., et al. Germ cell defects and hematopoietic hypersensitivity to gamma- interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996;88:49–58. [PubMed] [Google Scholar]
- 39.Kawabata T., Yamaguchi S., Buske T., Luebben S., Wallace M., Matise I., Schimenti J., Shima N. A reduction of licensed origins reveals strain-specific replication dynamics in mice. Mamm. Genome. 2011;22:506–517. doi: 10.1007/s00335-011-9333-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luebben S.W., Kawabata T., Akre M.K., Lee W.L., Johnson C.S., O'Sullivan M.G., Shima N. Helq acts in parallel to Fancc to suppress replication-associated genome instability. 2013;40:10283–10297. doi: 10.1093/nar/gkt676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lukas C., Savic V., Bekker-Jensen S., Doil C., Neumann B., Solvhoj Pedersen R., Grofte M., Chan K.L., Hickson I.D., Bartek J., et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011;13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- 42.Harrigan J.A., Belotserkovskaya R., Coates J., Dimitrova D.S., Polo S.E., Bradshaw C.R., Fraser P., Jackson S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011;193:97–108. doi: 10.1083/jcb.201011083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baumann C., Körner R., Hofmann K., Nigg E.A. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell. 2007;128:101–114. doi: 10.1016/j.cell.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 44.Carreau M. Not-so-novel phenotypes in the Fanconi anemia group D2 mouse model. Blood. 2004;103:2430. doi: 10.1182/blood-2003-11-3946. [DOI] [PubMed] [Google Scholar]
- 45.Wang L.C., Stone S., Hoatlin M.E., Gautier J. Fanconi anemia proteins stabilize replication forks. DNA Repair. 2008;7:1973–1981. doi: 10.1016/j.dnarep.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schlacher K., Wu H., Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaudhury I., Sareen A., Raghunandan M., Sobeck A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 2013;41:6444–6459. doi: 10.1093/nar/gkt348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zou L., Elledge S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 49.Byun T.S., Pacek M., Yee M.-C., Walter J.C., Cimprich K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 51.Chuang C.-H., Wallace M.D., Abratte C., Southard T., Schimenti J.C. Incremental genetic perturbations to MCM2-7 expression and subcellular distribution reveal exquisite sensitivity of mice to DNA replication stress. PLoS Genet. 2010;6:e1001110. doi: 10.1371/journal.pgen.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergoglio V., Boyer A.-S., Walsh E., Naim V., Legube G., Lee M.Y.W.T., Rey L., Rosselli F., Cazaux C., Eckert K.A., et al. DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013;201:395–408. doi: 10.1083/jcb.201207066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naim V., Wilhelm T., Debatisse M., Rosselli F. ERCC1 and MUS81–EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013;15:1008–1015. doi: 10.1038/ncb2793. [DOI] [PubMed] [Google Scholar]
- 54.Casper A.M., Nghiem P., Arlt M.F., Glover T.W. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 55.Fenech M. Cytokinesis-block micronucleus cytome assay. Nat. Protocols. 2007;2:1084–1104. doi: 10.1038/nprot.2007.77. [DOI] [PubMed] [Google Scholar]
- 56.Freie B., Li X., Ciccone S.L.M., Nawa K., Cooper S., Vogelweid C., Schantz L., Haneline L.S., Orazi A., Broxmeyer H.E., et al. Fanconi anemia type C and p53 cooperate in apoptosis and tumorigenesis. Blood. 2003;102:4146–4152. doi: 10.1182/blood-2003-03-0971. [DOI] [PubMed] [Google Scholar]
- 57.Wallace M.D., Southard T.L., Schimenti K.J., Schimenti J.C. Role of DNA damage response pathways in preventing carcinogenesis caused by intrinsic replication stress. 2013. [DOI] [PMC free article] [PubMed]
- 58.Niedzwiedz W., Mosedale G., Johnson M., Ong C.Y., Pace P., Patel K.J. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol. Cell. 2004;15:607–620. doi: 10.1016/j.molcel.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 59.Mirchandani K.D., McCaffrey R.M., D’Andrea A.D. The Fanconi anemia core complex is required for efficient point mutagenesis and Rev1 foci assembly. DNA Repair. 2008;7:902–911. doi: 10.1016/j.dnarep.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim H., Yang K., Dejsuphong D., D’Andrea A.D. Regulation of Rev1 by the Fanconi anemia core complex. Nat. Struct. Mol. Biol. 2012;19:164–170. doi: 10.1038/nsmb.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lossaint G., Larroque M., Ribeyre C., Bec N., Larroque C., Décaillet C., Gari K., Constantinou A. FANCD2 binds MCM proteins and controls replisome function upon activation of S phase checkpoint signaling. Mol. Cell. 2013;51:678–690. doi: 10.1016/j.molcel.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 62.Ying S., Minocherhomji S., Chan K.L., Palmai-Pallag T., Chu W.K., Wass T., Mankouri H.W., Liu Y., Hickson I.D. MUS81 promotes common fragile site expression. Nat. Cell Biol. 2013;15:1001–1007. doi: 10.1038/ncb2773. [DOI] [PubMed] [Google Scholar]
- 63.Parmar K., D’Andrea A., Niedernhofer L.J. Mouse models of Fanconi anemia. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2009;668:133–140. doi: 10.1016/j.mrfmmm.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tischkowitz M., Winqvist R. Using mouse models to investigate the biological and physiological consequences of defects in the Fanconi anaemia/breast cancer DNA repair signalling pathway. J. Pathol. 2011;224:301–305. doi: 10.1002/path.2903. [DOI] [PubMed] [Google Scholar]
- 65.Hughes C.R., Guasti L., Meimaridou E., Chuang C.-H., Schimenti J.C., King P.J., Costigan C., Clark A.J.L., Metherell L.A. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J. Clin. Invest. 2012;122:814–820. doi: 10.1172/JCI60224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gineau L., Cognet C., Kara N., Lach F.P., Dunne J., Veturi U., Picard C., Trouillet C., Eidenschenk C., Aoufouchi S., et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Invest. 2012;122:821–832. doi: 10.1172/JCI61014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hendzel M.J., Wei Y., Mancini M.A., Van Hooser A., Ranalli T., Brinkley B.R., Bazett-Jones D.P., Allis C.D. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]