


Abstract
Alternative pre-messenger ribonucleic acid (pre-mRNA) splicing is an essential process in eukaryotic gene regulation. The T-cell intracellular antigen-1 (TIA-1) is an apoptosis-promoting factor that modulates alternative splicing of transcripts, including the pre-mRNA encoding the membrane receptor Fas. TIA-1 is a multi-domain ribonucleic acid (RNA) binding protein that recognizes poly-uridine tract RNA sequences to facilitate 5′ splice site recognition by the U1 small nuclear ribonucleoprotein (snRNP). Here, we characterize the RNA interaction and conformational dynamics of TIA-1 by nuclear magnetic resonance (NMR), isothermal titration calorimetry (ITC) and small angle X-ray scattering (SAXS). Our NMR-derived solution structure of TIA-1 RRM2–RRM3 (RRM2,3) reveals that RRM2 adopts a canonical RNA recognition motif (RRM) fold, while RRM3 is preceded by an non-canonical helix α0. NMR and SAXS data show that all three RRMs are largely independent structural modules in the absence of RNA, while RNA binding induces a compact arrangement. RRM2,3 binds to pyrimidine-rich FAS pre-mRNA or poly-uridine (U9) RNA with nanomolar affinities. RRM1 has little intrinsic RNA binding affinity and does not strongly contribute to RNA binding in the context of RRM1,2,3. Our data unravel the role of binding avidity and the contributions of the TIA-1 RRMs for recognition of pyrimidine-rich RNAs.
INTRODUCTION
T-cell intracellular antigen-1 (TIA-1) is a ribonucleic acid (RNA)-binding protein that is implicated in alternative splicing regulation of various pre-messenger ribonucleic acid (pre-mRNA), with diverse functions of the proteins encoded such as in sex determination in Drosophila or apoptosis, homeostasis and survival of motor neurons in humans (1–3). Besides its role in alternative splicing, TIA-1 is able to mediate and suppress mRNA translation by binding to 3′ untranslated regions marked by AU-rich elements (AREs) in the cytoplasm during environmental stress (4–6). More recently, TIA-1 has also been implicated in translational control via binding to 5′ terminal oligopyrimidine-tract (5′ TOP) RNAs (7–8).
TIA-1 has been identified as an apoptosis-promoting factor that mediates programmed cell death by associating with the Fas-activated serine/threonine kinase (FAST-K) signal pathway (9). TIA-1 promotes the inclusion of exon 6 during splicing of human FAS pre-mRNA, which encodes a transmembrane protein that triggers programmed cell death. Alternative splicing by skipping of FAS exon 6 results in a soluble anti-apoptotic form of the Fas protein that is linked to autoimmune responses (10) and systemic lupus erythematosus (11). Pre-mRNAs targeted for splicing regulation by TIA-1 harbor short uridine-rich (U-rich) stretches located downstream of 5′ splice sites (12–15). Upon binding to these U-rich RNA motifs the TIA-1/RNA complex promotes 5′ splice site recognition by U1 small nuclear ribonucleoprotein (snRNP) through the interaction with the U1 snRNP-associated protein U1C (12). Phosphorylation of TIA-1 by FAST-K has been shown to enhance the recruitment of U1 snRNP to promote the inclusion of FAS exon 6 (16).
TIA-1 and its close homolog TIA-1-related protein, TIAR, have distinct expression patterns in different human tissues and cell lines (17–19). Both proteins are widely expressed in cells and mediate splicing of their own pre-mRNA (19–20). Two human TIA-1 isoforms, TIA-1a and TIA-1b, show similar sub-cellular distribution and RNA binding activity. The TIA-1a isoform has a 11 amino acid insertion preceding RRM2, corresponding to a 386 residue protein. TIA-1b lacks this insertion and displays enhanced splicing stimulatory activity compared to TIA-1a both in vitro and in vivo (19). As the biological role of TIA-1b has been well characterized by previous studies the present study focuses on TIA-1b, which will be referred to as TIA-1 from now on. TIA-1 encompasses 375 amino acids and consists of three RNA recognition motifs (RRM1, RRM2 and RRM3) and a C-terminal glutamine-rich domain (Q-rich domain) (Figure 1A and B), and the sequence alignment from different species are compared and aligned by ClustalW2 (21) and by ESPript (22–23). In vitro binding studies indicate that the three TIA-1 RRM domains have different capacities to bind RNA (12,17,24–25). The central TIA-1-RRM2 exhibits highest binding affinity towards pre-mRNA targets but RNA binding is further enhanced by the presence of RRM3. A recent study suggested a potential pH-dependent regulation of binding to AU-rich RNAs involving a histidine residue in an N-terminal extension of RRM3 (25). In contrast, TIA-1-RRM1 is thought to assist the Q-rich domain to recruit the U1 snRNP-associated protein U1C by protein–protein interactions (12).
Figure 1.
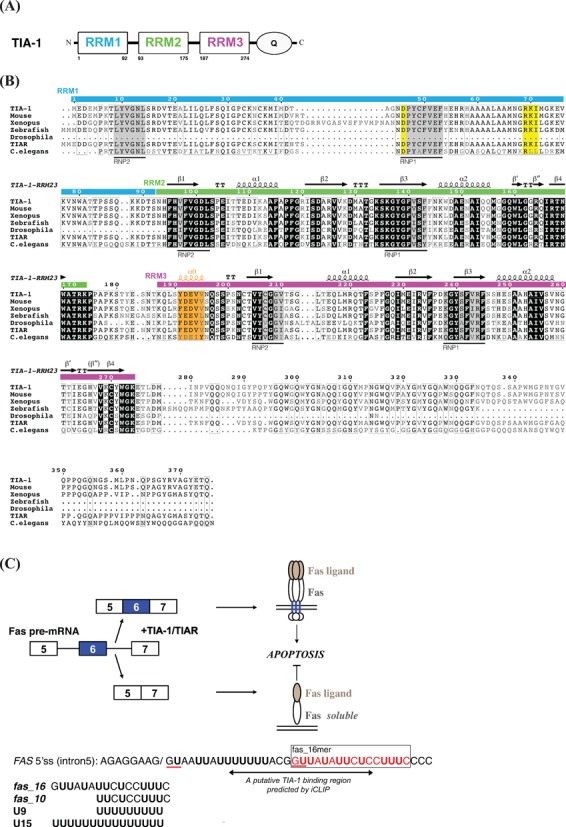
(A) Schematic representation of human TIA-1 domains. (B) Sequence alignment of TIA-1 and TIAR from different species. Original sequences used in the analysis: residues 1–375 of Homo sapiens TIA-1, residues 1–377 of Mus musculus TIA-1, residues 41–427 of Xenopus laevis TIA-1, residues 1–342 of Danio rerio (zebrafish) TIA-1, residues 166–356 of Drosophila melanogaster TIA-1, residues 1–375 of Homo sapiens TIAR and residues 45–373 of Caenorhabditis elegans TIAR are compared and aligned by ClustalW2 (21) and plotted by ESPript 2.2 (22,23). The secondary structure indicated corresponds to the solution structure of human TIA-1-RRM2,3. The RNP1 and RNP2 motifs are highlighted with gray background, the negatively charged residue in RNP1 and the RXI/V/L motif of RRM1 indicative of a potential UHM domain are shown with yellow background. The residues in RRM3 helix α0 are colored orange. (C) Alternative splicing of human FAS exon 6. The putative TIA-1 binding region in FAS intron 5 indicated by a double-arrow is predicted by iCLIP analyses, i.e. 10–28 nucleotides downstream of exon/intron boundaries. The GU motifs corresponding to 5′ splice sites in FAS intron 5 RNA are underlined in red. RNA sequences of the 16-mer pre-RNA ligand in FAS intron5 (fas_16) and a uridine-rich RNA (U9), used in the present study are indicated.
Recently, individual-nucleotide resolution UV-crosslinking and immunoprecipitation (iCLIP) experiments have mapped the regions of TIA-1 binding to pre-mRNA transcripts. The most frequent TIA-1 binding sites are observed 10–28 nucleotides downstream of exon/intron boundaries (26). A corresponding region downstream of the 5′ splice site of FAS intron 5 is thus relevant for studying the protein–RNA interaction of TIA-1 (Figure 1C). The presence and interactions with U1-C and the base pairing of the U1 snRNA with the 5′ splice site likely influences the binding of TIA-1 to the pre-mRNA. Not only uridine-rich stretches downstream but also upstream of an exon mediate TIA-1 binding (2).
TIA-1 is a typical multi-domain RNA binding protein where individual RNA binding domains connected by flexible linkers cooperate in the recognition of target RNAs. Understanding the contributions and interplay of the individual domains is required to dissect the molecular mechanisms of TIA-1 in the regulation of the Fas signal transduction pathway and apoptosis. So far, only experimentally determined structures of the unbound TIA-1 RRM2 domain have been reported by crystallography and NMR (27–28), which showed that RRM2 adopts a canonical RRM fold. The relative contributions and arrangements of the three RRM domains and their role for RNA recognition are not known. Using isothermal titration calorimetry (ITC) experiments, Kielkopf et al. have recently demonstrated that FAS intron 5 RNA exhibits two binding sites with comparable affinities for TIA-1, and proposed that all three RRMs contribute to high affinity binding to a poly-uridine RNA (U20) (24). However, the sequence motifs recognized in the FAS intron 5 RNA and molecular details are unknown.
Here, we have combined solution state NMR, ITC and small angle X-ray scattering (SAXS) experiments to study the structure, dynamics and RNA binding of TIA-1. NMR relaxation data and chemical shift comparisons indicate that, in the absence of RNA, the three RRMs of TIA-1 are independent structural modules connected by flexible linkers. The solution structure of a tandem RRM2,3 construct of TIA-1 (TIA-1-RRM2,3) reveals that RRM3 is unusual as it represents a canonical RRM fold that is extended by an N-terminal helix α0, consistent with previous secondary structure analyses (29). While RRM2 and RRM3 tumble independent from each other in the absence of RNA a uniform rotational correlation time is observed when bound to an RNA ligand derived from FAS pre-mRNA intron 5. SAXS analysis of RRM2,3 bound to this intron RNA ligand further demonstrates that the protein–RNA complex is rigid and compact—in contrast to the free protein. A similar compaction of the overall domain arrangements is also found for RRM1,2,3 upon binding to a U15 RNA ligand. NMR and ITC experiments reveal that TIA-1-RRM2,3 has two binding sites for a 16-mer FAS intron RNA and binds with 1:1 stoichiometry and nanomolar affinity to a 10-mer derived from the FAS intron RNA or to a U9 oligonucleotide. RRM1 has little intrinsic RNA binding affinity and does not strongly contribute to U15 RNA binding in the context of RRM1,2,3. Our results show that the multiple RRM domains of TIA-1 are structural independent modules in the absence of RNA, while RRM2,3 bind cooperatively to pyrimidine-rich RNA ligands.
MATERIALS AND METHODS
Sample preparation
Several TIA-1 constructs including RRM1 (residues 1–92), RRM2 (residues 93–175), RRM3 (residues 195–274), RRM2,3 (residues 93–274) and RRM1,2,3 (residues 1–274) were selected according to previously described studies (12). The amplified DNA fragments were sub-cloned into the expression vector pETtrx_1a vector (European Molecular Biology Laboratory, Heidelberg, Germany) between NcoI and KpnI sites, as a fusion with an N-terminal His-tag, a thioredoxin fusion tag and a TEV protease cleavage site. An N-terminal extension construct of RRM3 named RRM3 (residues 187–274) was made to have a complete RRM3 domain of TIA-1 by insertion into a modified pETM-11 vector, including an N-terminal His-tag, a Z-tag and separated by a TEV protease cleavage site. All recombinant proteins were transformed into Escherichia coli strain BL21 (DE3), except for RRM1, which was transformed into E. coli strain Rosetta (DE3) for expression. Unlabeled, 15N uniformly labeled, or 15N/13C uniformly labeled protein samples were expressed by growing cells in Luria–Bertani (LB) medium or M9 minimal medium (kanamycin 30 μg·ml−1) supplemented with 15NH4Cl and 13C-glucose at 20°C overnight after induction at OD600 of 0.6–0.8 with 0.5 mM IPTG. The cells were harvested by centrifugation and resuspended in buffer containing 50 mM sodium phosphate (pH 8.0) and 300 mM NaCl. The harvested cells were disrupted using a homogenizer (Microfluidics), and cell lysate and supernatant were separated by centrifugation at 45 000 × g for 30 min at 4°C. The supernatant protein was bound to Ni-NTA resin (Qiagen) and subsequently washed by buffer with 10 and 20 mM imidazole, respectively to remove most protein impurities, and then eluted with the same buffer containing 120 mM imidazole. Eluted protein was incubated with TEV protease at room temperature overnight for fusion tag removal. After buffer exchange to remove imidazole, a second Ni-NTA affinity column was applied to separate solubility tags from the tag free protein products. The tag free TIA-1 fragment was then purified by size-exclusion chromatography (HiLoad 16/60 Superdex 75, GE Healthcare Biosciences). The purity of the subsequent eluted protein was detected on a Coomassie-blue-stained sodium dodecyl sulfate (SDS) polyacrylamide gel (PAGE) and shown to be higher than 95%. RNA oligonucleotides used for NMR and ITC were purchased from Biospring GmbH, Frankfurt (Germany) and/or IBA GmbH, Göttingen (Germany). The final concentration of TIA-1 fragments and RNA were checked by absorbance at 280 and 260 nm, respectively, using a NanoDrop 1000 spectrophotometer (Thermo Scientific).
NMR spectroscopy
Spectra were acquired at 298 K using a AVIII500, AVIII600, AVIII750, AVIII800 and a AVI900 Bruker NMR spectrometer, equipped with cryogenic or room temperature (750 MHz) triple resonance gradient probes. Samples contained 0.1–0.7 mM protein in 10 mM sodium phosphate (pH 6.5), 50 mM NaCl, 10 mM DTT, 0.02% NaN3 with 10% 2H2O added for the lock. All spectra were processed using NMRPipe/Draw (30) and analyzed using Sparky (31) and NMRView (32). Protein backbone assignments for 15N, 1HN, 13Cα, 13Cβ and 13C′ chemical shifts were obtained from HNCACB, CBCA(CO)NH, HNCA and HNCO experiments (33). Amino acid side chain resonance assignments were obtained from standard HCCH-TOCSY, HBHA(CO)NH (34), 15N- and 13C-edited NOESY-HSQC experiments. Aromatic resonances were assigned using 2D 1H–13C HSQC, HBCBCGCDHD, HBCBCGCDCEHE (35) and NOESY spectra.
NMR relaxation measurements
To study the dynamical properties of protein free and bound with RNA, NMR data were recorded at 298 K for 0.15 mM of TIA-1-RRM1,2,3 and 0.55 mM 15N-labeled TIA-1-RRM2,3 protein free and bound to fas_16 RNA (ratio 1:2 of protein to RNA) on a 750 MHz NMR spectrometer at 298 K, while data for 0.2 mM 15N-labeled TIA-1-RRM1,2,3 bound with U15 RNA (ratio 1:1.2 of protein to RNA) were acquired on a 800 MHz Bruker NMR spectrometer. Amide 15N relaxation data of R1, R1ρ, and steady-state heteronuclear {1H}–15N-NOE experiments were performed as described (36–37). R1 data were measured with 10 different relaxation delays, 21.5, 86.5, 162, 248.4, 518.4, 669.6, 885.6, 1144.8, 1382.4 and 2160 ms, and R1ρ data were determined by using nine delay points of 5, 15, 25, 40, 55, 80, 110, 140 and 200 ms for RRM2,3, RRM2,3 bound to fas_16 RNA and free RRM1,2,3. For relaxation data recorded by RRM1,2,3 bound to U15: R1 data were measured with 13 different relaxation points and three redundant delays, 21.6/21.6, 43.2, 86.4, 172.8, 259.2, 345.6/345.6, 518.4, 669.6/669.6, 885.6, 1080, 1296, 1512 and 1728 ms, and R1ρ data were determined by using 10 different delay points together with two redundant delays of 5/5, 10, 15, 20, 30, 40, 50, 60, 80/80 and 100 ms. Duplicate time points were used for error estimation. The transverse relaxation rate R2 for each residue was estimated by correction of the observed relaxation rate R1ρ with the offset Δν of the rf field to the resonance using the relation R1ρ = R1 cos2θ + R2 sin2θ, where θ = tan−1(ν1/Δν). The correlation time (τc) of the protein molecule was then estimated using the ratio of averaged R2/R1 values (38). Steady-state heteronuclear {1H}–15N-NOE spectra were recorded with and without 3 s of 1H saturation. All relaxation experiments were acquired as pseudo-3D experiments and converted to 2D data sets during processing. The relaxation rates and error calculations were determined by using Sparky. The relaxation data of the resonances of Phe95–Leu102 in RRM1,2,3 could not be analyzed because the amide signals are not observed.
Small angle X-ray scattering experiments
30 μl of TIA-1-RRM2,3 (with and without fas_16 RNA) complex sample and buffer were measured at 25°C at the BioSAXS beamline BM29 at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France, using a 2D Pilatus detector. Ten frames with 2 s exposure time per frame were recorded for each complex and buffer sample, using an X-ray wavelength of λ = 1.008 Å. Measurements were performed in flow mode where samples were pushed through the capillary at a constant flow rate to minimize radiation damage. Frames showing radiation damage were removed prior to data analysis.
SAXS data reduction and integral parameters
For SAXS data collection and processing, the dedicated beamline software BsxCuBE was used in an automated fashion. The 1D scattering intensities of samples and buffers were expressed as a function of the modulus of the scattering vector Q = (4π/λ)sin θ with 2θ being the scattering angle and λ the X-ray wavelength. Buffer intensities were subtracted from the respective sample intensities using PRIMUS (39). The radii of gyration Rg of all samples were extracted by the Guinier approximation with the same program. The validity of the Guinier approximation, Rg for Q < 1.3, was verified and fulfilled in each case. Rg was also calculated as well as Dmax from pairwise distribution functions using GNOM (40). All statistics are summarized according to Jacques et al. (41) in Supplementary Table S1.
Structure calculation and validation
Automated assignments for the cross-peaks of 15N- and 13C-edited NOESY-HSQC spectra and structure calculations with torsion angle dynamics were done by using the program CYANA 3.0 (42). The set of NOE distance restraints derived from CYANA as well as the φ and ψ backbone dihedral angle restraints derived from TALOS+ (43) based on the chemical shifts were used for molecular dynamics simulated annealing and water refinement by using the program of RECOORD (44). The hydrogen bonding restraints are applied based on the identification of slow H/D exchange of amide protons by recording 1H,15N HSQC NMR spectra of lyophilized protein after redisolving in 100% D2O. All structures were validated by using iCing (45). Molecular images were generated with PyMol (Schrödinger) and MOLMOL (46).
NMR titrations
1H,15N HSQC spectra of TIA-1-RRM2,3 titrated with fas_16 and U9 RNA ligands were carried out to map the RNA-binding region of the complex. 0.1 mM 15N-labeled TIA-1-RRM2,3 was titrated with increasing amounts of RNA with various ratios of RNA to protein ranging from 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4 and 2.0 with fas_16, 0.05, 0.1, 0.2, 0.3, 0.4, 0.8 and 1.25 with U9, and 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4 and 2.0 with fas_10 RNA. Similarly, 0.1 mM of 15N-labeled TIA-1 RRM1,2,3 was titrated with increasing amounts of U15 RNA (0.1, 0.2, 0.4, 0.8 and 1.2 ratios). All measurements were acquired on a 600 MHz Bruker NMR spectrometer at 298 K using the NMR buffer described above. RNA oligonucleotides used for NMR titration were purchased and used directly by dissolving in a small amount of buffer without further purification. No pH change was observed for the protein sample after RNA addition.
Isothermal titration calorimetry
All titrations were carried out using MicroCal iTC200 calorimeters (Microcal, Northhampton, USA) at 25°C. To be consistent with the condition used for structural study, protein samples were dialyzed against NMR buffer without DTT to decrease noise. Buffer from the dialysis was used to resolubilize the RNA (BioSpring GmbH, Frankfurt, Germany) and to provide a baseline as required. The concentrations of 10 and 20 μM fas_16 and U9 RNA were used to be titrated with 150–200 μM TIA-1-RRM2,3, respectively. The reverse titrations were performed using 70–75 μM fas_16 and/or U9 RNA adding into 10 μM TIA-1-RRM2,3. Similar titrations were performed with RRM2,3 and fas_10 (concentrations used: 577 μM protein into 30 μM RNA and 58 μM protein with 300 μM RNA for reverse titration). For ITC measurements of RRM1,2,3 and U15, 300 μM U15 RNA were used to titrate into 30 μM TIA-1-RRM1,2,3, and the reversed titration was by ∼170 μM TIA-1-RRM1,2,3 adding into 15 μM U15 RNA. For each titration of RRM2,3 with fas_16 or U9, 26 injections of 1.5 ml of titrant were made at 180 s injection intervals, and data were collected at 25°C while stirring at 1000 revolutions per minute (rpm). Data were reduced with heat spikes from control experiments, where protein or RNA was titrated into buffer. Raw data were integrated and normalized using Origin ITC Analysis software provided by Microcal. Accordingly, a plot of ΔH per mole of injection versus molar ratio was produced and fit to an appropriate binding model (one-site or two-sites binding). Measurements have been repeated at least twice.
RESULTS
The three RRMs of TIA-1 are independent structural modules
A superposition of 1H,15N-HSQC spectra of the individual RRM domains and the 31 kDa RRM1,2,3 region of TIA-1 is shown in Figure 2A. The chemical shifts of the NMR signals in the individual RRMs are very similar compared with the RRM1,2,3 protein construct. Interestingly, several residues (Lys146, Asp148 and Glu150) in RRM2 helix α2 show significantly different chemical shifts in RRM1,2,3 when compared to the single RRM2 domain. These differences are not observed for the tandem RRM2,3 construct (Figure 2B). Charged residues (Lys88 and Lys89) in the RRM1–RRM2 linker also exhibit notable chemical shift differences in RRM1,2,3, suggesting that this linker may contact RRM2 helix α2 via charge–charge interactions. Except from these differences chemical shifts of the isolated domains and RRM1,2,3 are very similar. Also in the RRM2,3 tandem domain construct only very minor chemical shift variations are found (Ile111, Ala114, Phe118, Phe140 in RRM2 and Val210 in RRM3) when compared with the single domains (Figure 2B). These data indicate that the individual RRM domains in RRM2,3 and RRM1,2,3 are largely independent structural modules and lack strong domain–domain interactions. It is noteworthy that a few cross-peaks from a short RRM3 construct (residues 195–274, comprising the predicted canonical RRM secondary structure) have different chemical shifts when compared with the tandem RRM2,3 (Supplementary Figure S1). Secondary 13C chemical shifts (Supplementary Figure S2A) indicate that this region adopts an additional α-helix preceding residue 195 of the canonical RRM3 domain. Thus, the canonical RRM fold is extended by an additional helix and the fold of RRM3 and comprises residues 187–274, consistent with recent predictions (29). Average rotational correlation times (τc) were estimated for the individual domains from the trimmed mean ratio of 15N R2/R1 relaxation rates for RRM1,2,3 (Figure 2C), assuming isotropic rotational diffusion. This analysis shows similar τc values (∼12 ns) for RRM1 and RRM3 but significantly larger τc values (∼16 ns) for RRM2. A rotational correlation time of ∼15 ns would be expected for a 31 kDa protein, if the three RRMs would tumble together. Smaller τc values are expected if the individual RRMs do not interact strongly even though the rotational diffusion of the individual domains will still be coupled in spite of the presence of flexible linkers of 10 or more residues (47). The observation of distinct average τc values for residues in RRM2 compared with RRM1 and RRM3 and the virtual identical chemical shifts observed for the individual domains compared with the RRM1,2,3 multi-domain construct demonstrate that the individual RRMs tumble independently from each other and that there are no strong domain interactions in the free TIA-1 protein.
Figure 2.
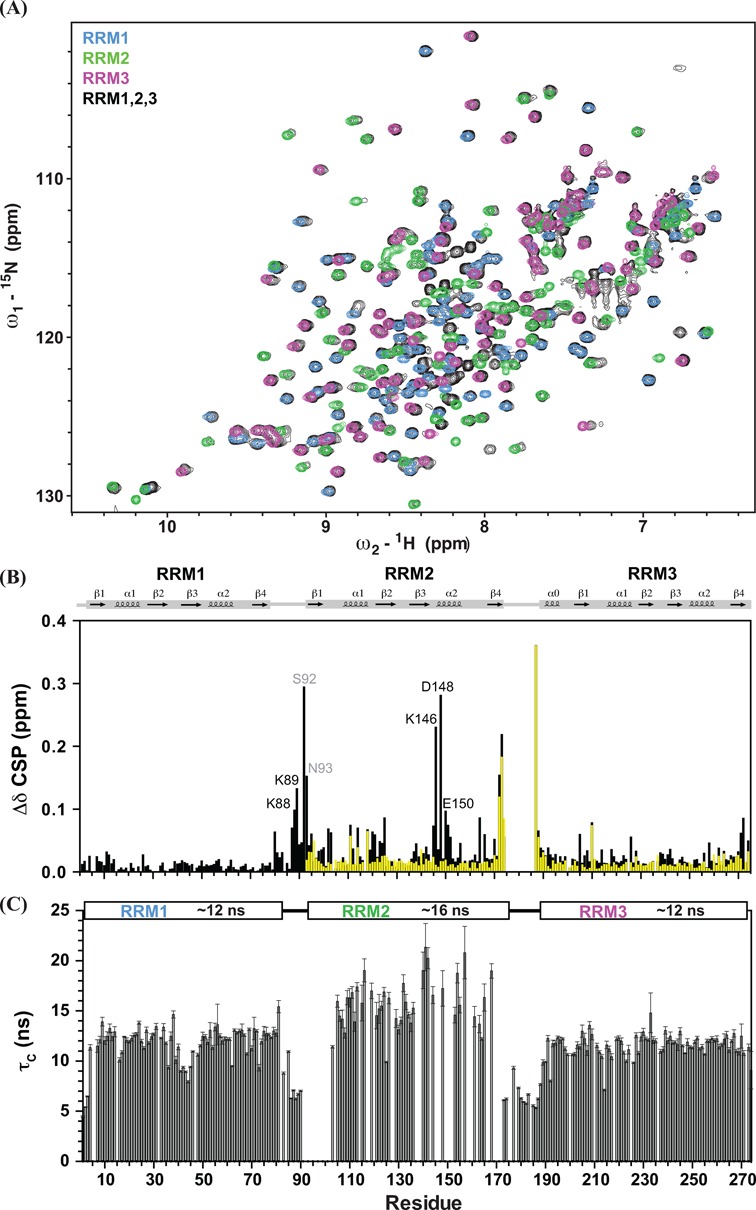
(A) The 1H,15N HSQC NMR spectra of individual RRM1 (blue), RRM2 (green), RRM3 (magenta) and RRM1,2,3 (black) are shown. (B) Differences of chemical shifts of individual RRM with RRM1,2,3 (black) and with RRM2,3 (yellow) are plotted versus residue number. Some distinct differences are observed in the RRM2 helix α2 (Lys146, Asp148 and Glu150) when compared with the resonances of RRM1,2,3. Residues in the RRM1–2 linker (Ser92 and Asn93) and two predicting residues (Lys88 and Lys89) also exhibit large chemical shift differences, suggesting that charge complementary interactions may contribute to the contacts of the RRM1–2 linker and RRM2. (C) Local rotational correlation times (τc) estimated from the ratio of 15N R2/R1 relaxation rates of RRM1,2,3 versus residue number are shown, with average τc values for each RRM domain indicated above. Error bars are derived from the relaxation data.
NMR relaxation data of the TIA-1 RRM2,3 tandem domains and virtually identical chemical shifts of the individual RRM2 and RRM3 domains compared with RRM2,3 indicate that also in this fragment the individual domains tumble independently with no significant domain–domain interactions between RRM2 and RRM3 in absence of RNA (see below).
Solution structure of TIA-1 RRM2,3
To understand the role of RRM3 in enhancing the RNA binding affinity of RRM2 (12), we determined the NMR-derived solution structure of the tandem TIA-1-RRM2,3 domains. The ensemble of the 10 lowest energy structures of TIA-1 RRM2,3 after water refinement is shown in Figure 3A, structural statistics are summarized in Table 1. As indicated by chemical shift analysis and NMR relaxation data the two domains do not interact, consistent with the absence of inter-domain NOEs. Therefore, the two RRM domains are presented and analyzed here individually (Figure 3B). RRM2 adopts the canonical βαββαβ RRM fold composed of a four-stranded anti-parallel β-sheet covered on one side by two α-helices. The structure of RRM2 in the RRM2,3 protein resembles that of the isolated TIA-1 RRM2 domain reported previously (27–28), with backbone coordinate RMSDs of 0.91 ± 0.08 and 0.81 ± 0.05 Å to the RRM2 structure determined by NMR (PDB: 2RNE) and crystallography (PDB: 3BS9), respectively. RRM3 in RRM2,3 superimposes with the structure of the isolated RRM3 of the homologous TIAR protein (PDB: 1X4G) with a backbone coordinate RMSD of 0.79 ± 0.06 Å. Notably, in RRM3, the canonical RRM fold is preceded by an additional α-helix located in the RRM2–RRM3 linker (helix α0, Tyr191–Val196), consistent with 13C secondary chemical shifts (Supplementary Figure S2A). Notably, helix α0 packs against RRM3 helix α2 and strand β4 and thus defines an enlarged RRM fold for RRM3. Helix α0 is not required for stability of RRM3, as a protein lacking helix α0 is stable and folded (Supplementary Figure S1), consistent with a recent report (29). The remaining part of the RRM2–RRM3 linker (Thr172–Ser190) in RRM2,3 does not form any secondary structure, consistent with random coil secondary chemical shifts and 15N relaxation data (Supplementary Figure S2A, see also below).
Figure 3.
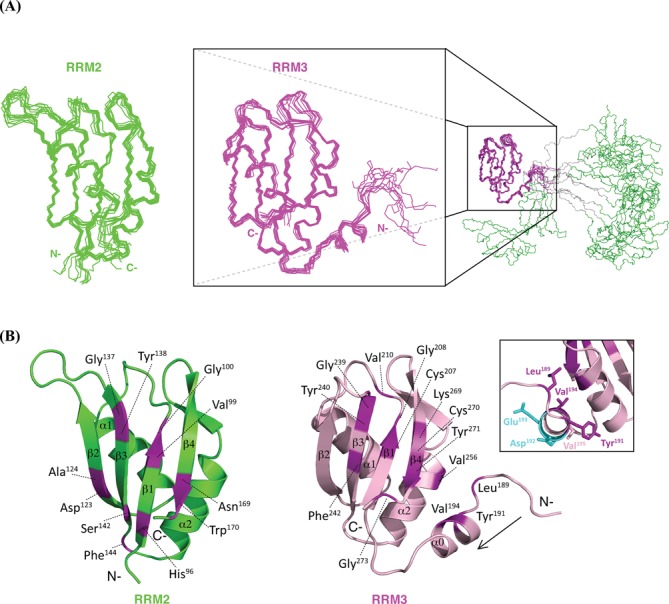
(A) The ensemble of the lowest 10 energy structures of TIA-1-RRM2,3 generated by superposition of backbone atoms of RRM2 (residue 93–173, left) and RRM3 (residue 184–274, right) domains, respectively. The insert illustrates that the two domains do not interact, with residues in the RRM2–RRM3 linker colored in gray. (B) TIA-1-RRM2,3 ribbon representation of RRM2 and RRM3. Residues with exhibit significant chemical shift perturbation upon titration of RRM2,3 with fas_16 RNA are colored purple and annotated (Δδ > 0.16 ppm, average chemical shift perturbation over all resonances with standard deviation ()). The CSPs reveal that for both RRM2 and RRM3 residues in the RNP2 (VFVGDL/VYCGGV) and RNP1 (KGYGFVSF/KGYSFVRF) motifs (β1 and β3 strands), as well as additional conserved residues in β4 are involved in RNA binding. The inset shows side chains of residues in RRM3 helix α0 (YEDVV) with the orientation along the arrow. Solvent exposed acidic side chains of Asp192 and Glu193 are colored in cyan. The side chains of Leu189, Tyr191 and Val194 pack against RRM3 helix α2.
Table 1. Structural statistics for TIA-1 RRM2,3.
| Structural statistics for TIA-1-RRM2,3a | |
|---|---|
| Structure calculation restraints | |
| Distance restraints | |
| Total NOEs | 2104 |
| Intra-residue | 485 |
| Sequential (|i − j| = 1) | 581 |
| Medium-range (|i − j| ≤ 4) | 395 |
| Long-range (|i − j| > 4) | 643 |
| Hydrogen bonds | 154 |
| Dihedral restraints (ϕ + ψ) | 272 |
| Quality analysis | |
| Restraints violations (mean ± SD) | |
| Distance restraints (Å) | 0.016 ± 0.00 |
| Dihedral angle restraints (°) | 0.687 ± 0.06 |
| Deviation from idealized geometry | |
| Bond length (Å) | 0.011 ± 0.00 |
| Bond angles (°) | 1.214 ± 0.03 |
| Impropers (°) | 1.387 ± 0.10 |
| Average pairwise r.m.s. deviation (Å)a | |
| Heavy (RRM2/RRM3) | 1.23 ± 0.15/0.88 ± 0.08 |
| Backbone (RRM2/RRM3) | 0.64 ± 0.17/0.41 ± 0.06 |
| Ramachandran values (%)a,b | |
| Most favored regions | 93.2 |
| Allowed regions | 6.8 |
| Generously allowed regions | 0 |
| Disallowed regions | 0 |
| WhatIf analysisa,c | |
| First generation packing | 2.590 ± 0.910 |
| Second generation packing | 7.285 ± 2.633 |
| Ramachandran plot appearance | −1.157 ± 0.170 |
| Chi-1/Chi-2 rotamer normality | −1.941 ± 0.517 |
| Backbone conformation | 0.408 ± 0.167 |
aFor residues 93–171 (RRM2) and 191–272 (RRM3)
bWith Procheck.
cAnalyzed by iCING. Structure Z-scores, a positive number is better than average.
RNA binding of the individual TIA-1 RRM domains
To characterize the RNA binding properties of the TIA-1 RRM domains to pyrimidine-rich ligands, NMR chemical shifts of amides were monitored upon titration of a U9 RNA oligonucleotide to the individual 15N-labeled TIA-1 RRM domains (Figure 4A–C). The stronger chemical shift perturbations (CSPs) (48) of RRM2 upon U9 binding compared to the RRM1 and RRM3 are in agreement with the previously reported higher affinity to uridine-rich RNAs (12,17,24). Notably, in addition to the two RNP motifs, the additional N-terminal helix α0 and C-terminal residues in RRM3 exhibit strong CSPs. This indicates that these regions are affected by binding to the U9 RNA either by engaging direct contacts to the RNA or by indirectly induced conformational changes. The reduced RNA binding of RRM3 reported previously is likely a result of the fact that these studies (12,17) have used a truncated version of RRM3 lacking helix α0.
Figure 4.
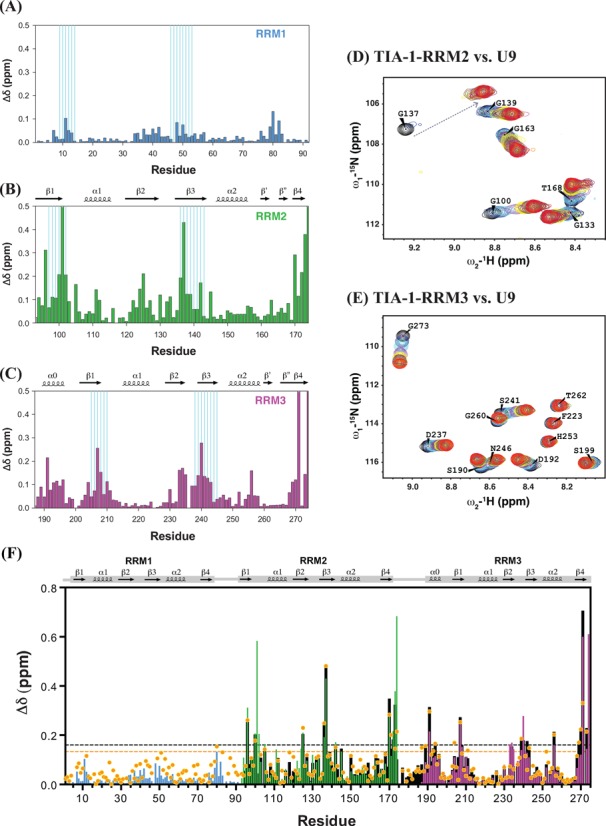
Chemical shift perturbations of individual TIA-1 RRM domains upon binding to U9 RNA. The combined 1H and 15N chemical shift perturbations were calculated using the equation: Δδ = [(ΔδHN)2 + (ΔδN/6.5)2]1/2 (48). The CSP of amide groups in (A) RRM1, (B) RRM2 and (C) RRM3 are plotted over residues, RNP motifs are indicated by cyan lines. Titration data are shown for zoomed regions of 1H, 15N HSQC spectra of (D) RRM2 and (E) RRM3. Spectra are colored as: free protein (black) and in the presence of U9 RNA RNA at 0.1 (blue), 0.2 (cyan), 0.4 (purple), 0.6 (yellow), 0.8 (brown), 1.0 (magenta), 1.2 (green), 1.4 (gray), 1.6 (orange) and 1.8 (dark-red/red) and 2.0 (red) molar equivalents. (F) Comparison of CSPs of tandem RRM2,3 (black), RRM1 (residues 1–92, in light blue), RRM2 (residues 93–174, in green) and RRM3 (residues 187–274, in magenta) upon binding to U9 RNA with TIA-1-RRM1,2,3 binding to U15 RNA (orange dots). The CSPs are plotted versus residue numbers. The dashed line in black shows a cut-off Δδ > 0.16 ppm (average chemical shift perturbation over all resonances + standard deviation) for RRM2,3 titrated with U9, and the one in orange for Δδ > 0.13 ppm for RRM1,2,3 with U15.
U9 RNA binding to the individual RRM3 domain is fast on the NMR chemical shift time scale (Figure 4E) as cross-peaks positions are shifting with increasing RNA concentration. Hence, RRM3 binds with a dissociation constant KD ∼30–50 μM as derived from the NMR titration (Supplementary information, Figure S3B). Binding kinetics observed with the NMR titrations of RRM2 are fast to intermediate on the NMR chemical shift time scale as some residues disappear and reappear with increasing U9 concentration (e.g. Gly137; Figure 4D). When analyzing residues with fast chemical exchange behavior an apparent KD ∼35 μM is obtained (Supplementary Figure S3A). The binding affinities observed are consistent with ITC measurements for the interaction of mouse TIA-1-RRM2 with a U5 RNA oligonucleotide with KD ∼30 μM (28). The observation that RRM2 binding kinetics is fast to intermediate in the NMR titrations indicates a stronger interaction of TIA-1-RRM2 to poly-U RNA compared with RRM3.
Similar chemical shift perturbations are observed for the single RRM2, RRM3 domains and the tandem RRM2,3 protein upon saturated binding to U9 RNA (Figure 4A–E, black bars in Figure 4F). This indicates that identical binding interfaces are employed by the isolated RRM domains even though the overall binding affinity of RRM2,3 is much higher due to cooperative effects (see below). Intriguingly, compared with RRM2 and RRM3 (Figure 4B and C), RRM1 exhibits only very minor chemical shift perturbations upon addition of U9 RNA (Figure 4A), which may reflect non-specific contacts due to its overall positively charged surface. To explore whether RRM1 could nevertheless contribute to high affinity RNA-binding in the RRM1,2,3 construct, we performed NMR titrations of RRM1,2,3 with an U15 RNA oligonucleotide. The chemical shift perturbations of RRM1,2,3 upon binding to U15 RNA (Figure 4F; Supplementary Figure S5A) are very similar for RRM2,3 but very small for RRM1. The CSPs observed for RRM1 within RRM1,2,3 are slightly stronger than seen for the RRM1/U9 RNA interaction but much smaller compared to the CSPs seen for RRM2 and RRM3 and thus do not indicate specific RNA contacts. Thus, RRM1 does not seem to mediate significant contacts with the RNA even in the context of RRM1,2,3. This is further confirmed by ITC data which suggest that RRM1 does not contribute to U15 RNA binding (Supplementary Figure S8 and see below).
Thus, RRM1 lacks significant intrinsic RNA binding activity, consistent with the presence of degenerate RNP motifs (that are normally important for RNA binding, Figure 1B) and might thus primarily be involved in mediating protein–protein interactions. Sequence identities of human TIA-1 compared to those of mouse, frog, zebrafish and human TIAR are 96, 74, 80 and 79%, respectively (49). Although the sequence identity is <35% between human and Drosophila, the RRM2,3 region shows over 60% homology, suggesting that the tandem TIA-1-RRM2,3 domains have evolutionary conserved functions in RNA recognition. In contrast, the lower conservation of RRM1 and the C-terminal Q-domain indicate a lower selection pressure for these regions, or that residues involved in RRM1-mediated protein–protein interactions have co-evolved in higher organisms only.
Binding of TIA-1-RRM2,3 to an intronic sequence in FAS pre-mRNA
Regulation of FAS alternative splicing (Figure 1C) requires binding of TIA-1 to pyrimidine-tracts located in the 5′ region of pre-mRNA introns. To characterize the RNA recognition with a ligand found in a biologically relevant substrate we analyzed the interaction of TIA-1 with 16-mer (fas_16) and 10-mer RNA (fas_10) oligonucleotides derived from FAS intron 5 (Figure 1C). This RNA comprises a GU di-nucleotide, resembling an alternative 5′ splice site followed by a strong Py-tract and is located 10–28 nt downstream of the exon/intron boundary of FAS exon 5. A poly-uridine oligonucleotide (U9) was used to compare TIA-1 binding to a general pyrimidine-rich ligand.
NMR titration of fas_16, fas_10, and U9 RNA to TIA-1-RRM2,3 shows binding in the intermediate to slow exchange regime at the NMR chemical shift time-scale, i.e. NMR signals of residues involved disappear and reappear at a different position upon saturation (Supplementary Figure S6). Therefore, to unambiguously determine the chemical shifts of RRM2,3 bound to fas_16 and fas_10 RNA backbone chemical shifts were re-assigned using triple-resonance experiments. The 13C secondary chemical shifts of the complex show that the secondary structure of RRM2,3 is not altered in the protein–RNA complex (Supplementary Figure S2B). Similar overall CSPs are observed for the amide signals of RRM2,3 upon either fas_16, fas_10 RNA or U9 binding (Figure 5A and B). This seems to reflect that all ligands are pyrimidine-rich and demonstrates that the same binding interface on RRM2,3 is employed for the recognition of these ligands. The binding surface with fas_16 is mapped onto a ribbon representation of TIA-1-RRM2,3 (Figure 3B) with significant CSP values (50). The interaction mainly involves the β-sheet surfaces of RRM2 and RRM3, similar to what has been seen for titrations of U9 RNA to the individual RRMs (Figure 4B and C), and corresponds to the canonical RNA binding surface of RRM domains. Notably, RRM3 helix α0 (Tyr191 and Val194) as well as residues that are in close spatial proximity (Leu189 preceding RRM3 helix α0 and Val256 in RRM3 helix α2, Figure 3B) are also strongly affected (Figure 5B).
Figure 5.
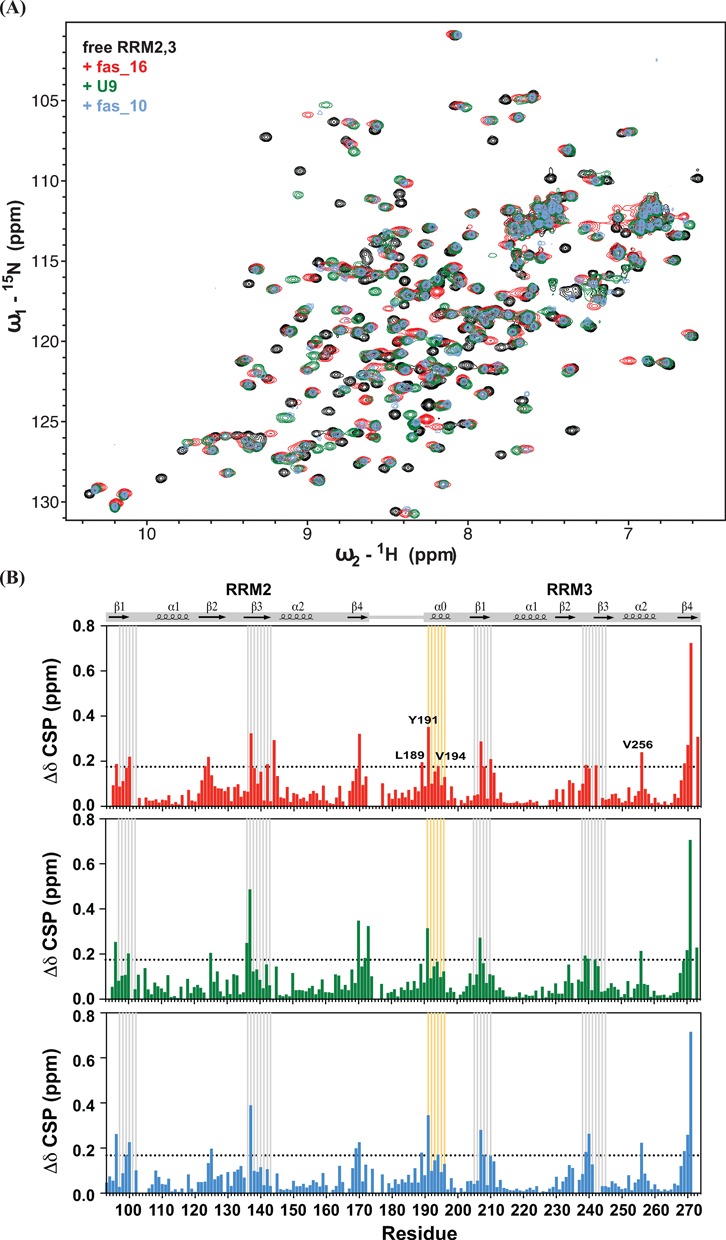
(A) Comparison of 1H,15N HSQC spectra of free RRM2,3 (black), and bound with fas_16 (red), U9 (green) and fas_10 (light blue) RNA. (B) The combined 1H and 15N chemical shift perturbations (Δδ) upon saturated binding by fas_16, U9, and fas_10 RNA are shown versus residue number. RNP motifs are indicated with gray lines. Residues of RRM3 helix α0 are highlighted by orange lines, and those with significant CSP are annotated.
We used ITC to determine the binding stoichiometry and affinity of TIA-1-RRM2,3 towards U9 and FAS intron RNA (Figure 6; Table 2). Upon titration of TIA-1-RRM2,3 to fas_16 RNA (Figure 1C) two distinct binding sites are observed with KD1 = 0.35 nM and KD2 = 27 μM. In a reverse titration (RNA into protein) an average dissociation constant of KD = 22 μM with 2:1 protein:RNA stoichiometry is found (Figure 6A). Upon titration of RRM2,3 into U9 RNA and vice versa dissociation constants of KD = 7.0 and 5.6 nM are determined, respectively (Figure 6B). Negative binding enthalpies ΔH observed for these ligands (Table 2; Supplementary Figure S7) indicate that the RNA binding is enthalpically driven.
Figure 6.
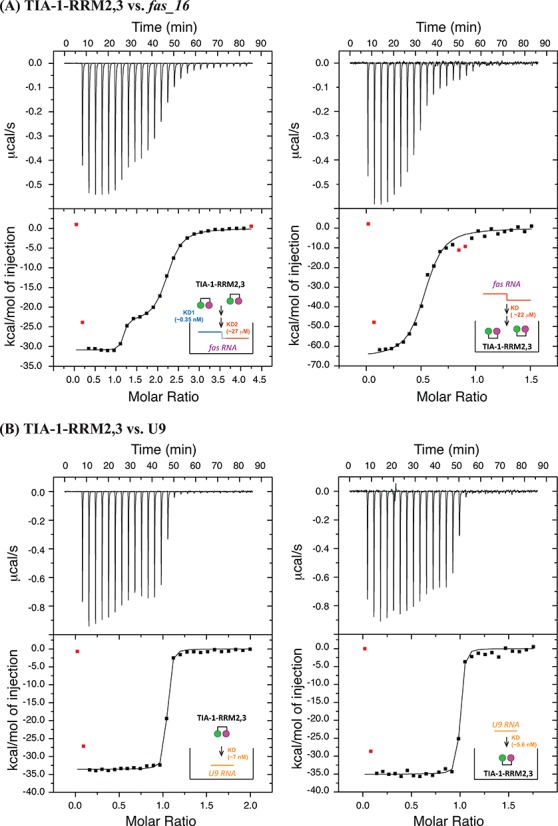
Isothermal titration calorimetry for the binding of TIA-1-RRM2,3 to fas_16 and U9 RNAs. (A) 10 μM protein (in the calorimetric cell) was titrated with 70 μM fas_16 (in the syringe), and the reverse titration by using TIA-1-RRM2,3 (200 μM) as titrants into RNA titrate (10 μM). (B) 20 μM protein was titrated with 200 μM U9 RNA, and the reverse titration of protein to U9 with the same concentrations. Each plot shows the heat released versus time in the upper panel with a thermogram of the integrated peak intensities plotted against the molar ratio of the complex in the lower panel. Fitted thermodynamic parameters data for each titration were shown in Table 2. All experiments were conducted in 10 mM sodium phosphate (pH 6.0) with 50 mM NaCl at 25°C.
Table 2. Isothermal titration calorimetry data for RNA binding of TIA-1 RRM2,3.
| KA (M−1) | KD (nM) | ΔG (kcal /mol) | ΔH (kcal /mol) | ΔS (cal /K·mol) | N | |
|---|---|---|---|---|---|---|
| RRM2,3 → fas_16 | ||||||
| Site 1 | (2.9 ± 1.0) × 109 | 0.4 ± 0.1 | −12.9 ± 0.2 | −30.9 ± 0.1 | −60.4 ± 0.7 | 1.1 |
| Site 2 | (3.7 ± 0.2) × 106 | 272.5 ± 14.9 | −9.0 ± 0.0 | −23.9 ± 0.2 | −50.1 ± 0.1 | 1.1 |
| fas_16 → RRM2,3 | (4.6 ± 1.0) × 106 | 216.0 ± 47.6 | −9.1 ± 0.1 | −61.3 ± 2.1 | −175.0 ± 0.5 | 0.5 |
| RRM2,3 → U9 | (1.4 ± 0.3) × 108 | 7.0 ± 0.2 | −11.1 ± 0.0 | −33.6 ± 0.2 | −75.5 ± 0.1 | 1.0 |
| U9 → RRM2,3 | (1.8 ± 0.6) × 108 | 5.6 ± 1.9 | −11.3 ± 0.2 | −35.1 ± 0.3 | −80.0 ± 0.7 | 1.0 |
| RRM2,3 → fas_10 | (2.8 ± 0.6) × 107 | 35.6 ± 8.1 | −10.2 ± 0.4 | −31.3 ± 0.3 | −71.1 ± 0.2 | 1.0 |
| RRM1,2,3 → U15 | (6.0 ± 0.9) × 107 | 16.9 ± 2.7 | −10.6 ± 0.1 | −42.4 ± 0.4 | −106.8 ± 1.1 | 1.0 |
The fitting model used: 2-site for RRM2,3 titrated into fas_16, 1-site for all other titrations. The change of Gibbs free energy and entropy were calculated using the equations: −TΔS = ΔG − ΔH and ΔG = −RT ln(KA) at T = 298 K.
Interestingly, the binding stoichiometry of TIA-1-RRM2,3 to fas_16 is 2:1 but 1:1 for U9. The presence of a high affinity binding site in fas_16 RNA with >10-fold increased binding affinity compared to U9 suggests either the specific recognition of a sequence motif in fas_16 RNA or avidity effects in binding to a longer pyrimidine-rich stretch, as commonly only 10–12 nucleotides are recognized by tandem RRMs. Within fas_16 RNA a 10-mer sequence motif may be sufficient for binding to RRM2,3, while the additional nucleotides present in the 16-mer ligand may contribute to avidity by binding in multiple registers and increased local concentrations of the pyrimidine-rich ligand. In an attempt to identify a putative minimal RNA sequence motif within fas_16 RNA we studied RRM2,3 binding to fas_10, which corresponds to the pyrimidine-rich region at the 3′-end of fas_16 (Supplementary Figrue S8A). Upon titration of RRM2,3 into fas_10 RNA, a dissociation constant of KD ∼17 nM was determined with a 1:1 stoichiometry, thus comparable to U9 RNA binding affinity. This suggests that the stronger binding to fas_16 mainly results from avidity effects upon binding to a longer pyrimidine-rich RNA ligand and that the RNA binding specificity of TIA-1 mainly requires pyrimidine-rich sequence.
In order to compare the contribution of RRM1 in RRM1,2,3 for RNA binding, we carried out ITC titrations of RRM1,2,3 with U15 RNA (Supplementary Figrue S8B). These experiments show KD values of 16 nM assuming a 1:1 binding event. Thus, the binding affinity of RRM1,2,3 to U15 RNA is similar to that of RRM2,3 to U9. This finding confirms the NMR chemical shift perturbation data which do not suggest strong contributions of RRM1 to RNA binding and suggest that RRM2,3 represent the high-affinity RNA binding region of TIA-1.
The TIA-1-RRM2,3–RNA complex is compact
To study internal motion and characterize the overall domain dynamics we measured 15N relaxation data for TIA-1 RRM2,3 in the RNA free and bound state (Figure 7A). The average tumbling correlation time for residues in RRM3, τc ≈ 10 ns, resembles the value expected for a molecular weight of 20.4 kDa whereas the average τc of RRM2 of 12 ns is slightly higher. These values suggest that the rotational diffusion of both domains is coupled, as the correlation times expected for the individual RRM2/RRM3, τc ≈ 6.1 ns/5.9 ns, estimated using HYDRONMR (51) are significantly lower than the experimentally derived values in context of the tandem RRM2,3. The presence of motional coupling in multi-domain proteins has been found even in the presence of a relatively long flexible linker connecting two domains (47) to be observed, as is the case for RRM2,3 (Figure 7A). However, the different values determined for the average tumbling correlation times of the two domains suggest that RRM2 and RRM3 do not interact (Figure 7B). The higher values observed for RRM2 (in spite of its smaller molecular weight) presumably reflects non-specific aggregation of this domain, as is also found for the RRM1–RRM2–RRM3 fragment (Figure 2C).
Figure 7.
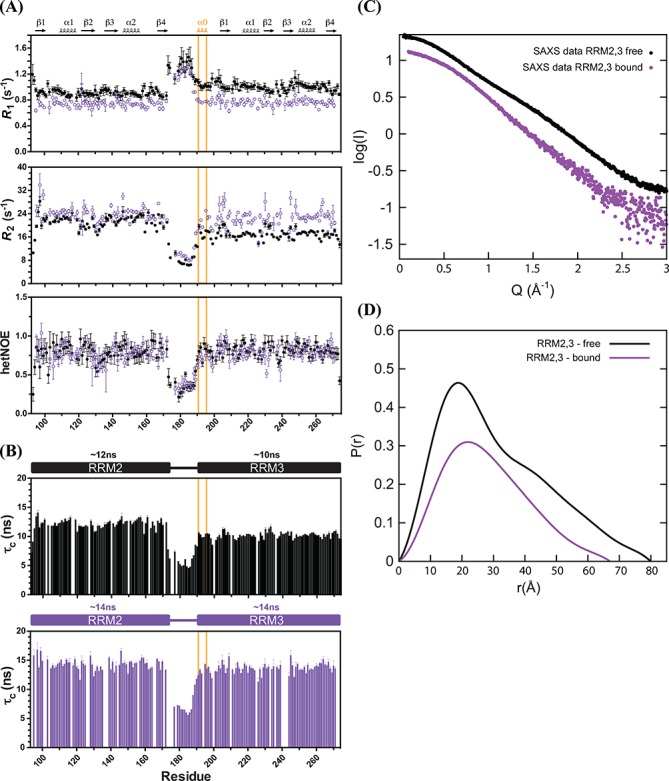
Relaxation and SAXS data of free and fas_16 RNA bound TIA-1-RRM2,3. (A) R1 (top), R2 (derived from R1ρ, middle) and {1H}-15N heteronuclear NOE values (bottom) of free (closed circles in black) and bound form (open circles in purple) measured at 298 K and pH 6.0 are shown. The average rotational correlation times (τc) for RRM2 and RRM3 in the RRM2,3 protein free (black) and bound to fas_16 (purple) are compared in (B). Residues in RRM3 helix α0 are highlighted with an orange box. (C) SAXS data of TIA-1-RRM2,3 free (black) and bound to fas_16 RNA (purple) and (D) the corresponding pairwise distribution functions.
Upon binding to fas_16 RNA both RRMs exhibit the same average tumbling correlation time of τc ≈ 14 ns, a value that is also consistent with the molecular weight of the complex (25.5 kDa). {1H}–15N heteronuclear NOE values demonstrate that the RRM2–RRM3 linker (residues 171–190) remains highly flexible and does not become ordered upon RNA-binding, consistent with the lack of chemical shift perturbations in the linker region upon RNA binding. Thus, RNA binding by TIA-1 RRM2,3 induces a compact domain arrangement where both domains cooperate in binding to the RNA ligand. We also determined the average tumbling correlation time of RRM1,2,3 bound with U15 (∼36 kDa). Although τc values for RRM2 (∼18 ns) are still slightly larger than for RRM1 (∼16 ns) or RRM3 (∼16.5 ns) (Supplementary Figrue S5B), the overall similar values suggests that in the RRM1,2,3/U15 RNA complex the three RRMs tumble together.
To provide additional support for the formation of a compact protein–RNA complex we recorded SAXS data for free and fas_16 RNA bound TIA-1 RRM2,3 (Figure 7C). Upon RNA binding the radius of gyration, Rg, decreases from 24.1 ± 0.1 to 22.0 ± 0.26 Å based on the Guinier approximation or 24.4 ± 0.003 to 21.6 ± 0.003 Å as calculated from the pairwise distribution function (Figure 7D). The maximum pairwise distance, Dmax, decreases from 80 Å in the absence of RNA to 67 Å upon RNA binding (SAXS statistics according to Jacques et al. (41) are listed in Supplementary Table S1). The larger Rg for TIA-1 RRM2,3 in the absence of RNA confirms that the two RRMs have no fixed orientation towards each other. The averaged ensemble is therefore larger in diameter than in the presence of RNA, where the Rg decreases substantially.
DISCUSSION
Here, we have identified RRM2 and RRM3 as the bona fide RNA binding domains of TIA-1 and report the high-resolution structures of RRM2 and RRM3 within a protein comprising the tandem TIA-1 RRM2,3. In the absence of RNA, the three RRMs of TIA-1 tumble independently as shown by NMR relaxation data and the lack of observable inter-domain NOEs in RRM2,3. The same is true for RRM2 and RRM3 within the TIA-1 RRM2,3 tandem domain construct, where NMR relaxation data indicate that the two domains tumble independently. However, we cannot exclude that weak and transient contacts between RRM2 and RRM3 may exist as has been suggested in a recent study where the presence of RRM2 was found to thermodynamically stabilize RRM3 in thermal unfolding experiments (29). In any case, our NMR data clearly indicate that both domains cooperate in binding to RNA ligands. When bound to RNA, both RRM domains exhibit a uniform tumbling rate, suggesting a synergistic effect in RNA binding by TIA-1-RRM2,3 and the formation of a compact protein–RNA complex. This is corroborated by SAXS measurements, where the gyration radius of RNA bound RRM2,3 is decreased by 12% compared with free RRM2,3.
We found that RRM3 represents a novel extended RRM structure where the canonical RRM fold is preceded by an N-terminal helix α0. The presence of an N-terminal helix has been proposed recently (29) and is also observed in the RRM3 of TIAR (PDB: 1X4G), indicating that this is a conserved feature of TIA proteins (Figure 8A and B). It has been previously speculated that this helix does not contribute additional RNA contacts (25). However, our data clearly indicate that RRM3 helix α0 plays a critical role in RNA recognitions, as residues in this helix experience almost the strongest chemical shift perturbations upon RNA binding. Interestingly, the strong chemical shift perturbations for RRM3 helix α0 upon RNA binding have not been observed in a recent study with the isolated RRM3 domain (25). Although in this study an AU-rich RNA ligand was used, this suggests that TIA-1 RNA binding depends on cooperative interactions involving both RRM2 and RRM3. Notably, His248 in helix α0 and additional histidine residues located within or near the RNA binding surface may play a role for the suggested pH-dependence of RNA binding by TIA-1 RRM3, even though these effects have not been confirmed in larger constructs of TIA-1 (25). Other structural extensions and variations of the canonical RRM fold have been reported recently (52–53). Comparison of TIA-1-RRM3 with other non-canonical RRM domains indicates that their helical extensions can also mediate additional RNA contacts, as for example seen in the Cst64 RRM (Figure 8C) (54). In contrast the C-terminal RRM domain of the human La protein shields the β-sheet surface and thus blocks RNA binding (Figure 8D) (55).
Figure 8.
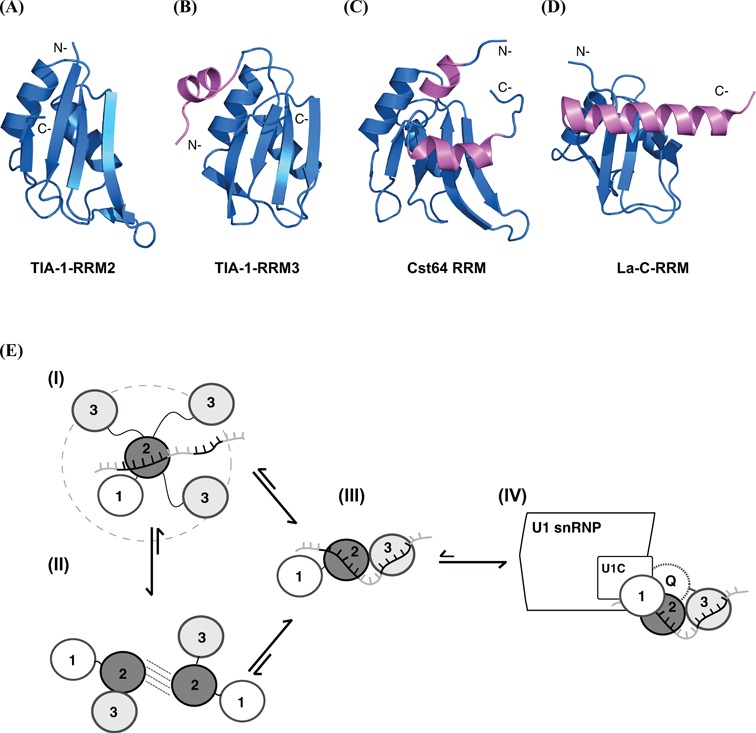
Comparison of the RRM domains of (A) TIA-1 RRM2, (B) TIA-1 RRM3, (C) Cst64 RRM (1P1T) (54) and (D) La-C-RRM (1OWX) (55). N- or C-terminal helical extensions are colored in magenta. (E) A model for RNA and protein interactions by TIA-1 RRM2,3. In free RRM1,2,3 individual RRM domains tumble independently. RRM2 mediates initial contacts to an RNA ligand followed by fly-casting of RRM3 for formation of a stable and rigid RRM2,3–RNA complex (I). Although there are no strong and specific domain–domain contacts detected in free RRM2,3, transient/pre-existing domain–domain interactions between RRM2 and RRM3 may contribute to speed up RNA recognition and skipping the fly casting step as shown in (II). The RRM1 and Q-domains mainly contribute to U1 snRNP recruitment by mediating protein–protein interactions.
It seems plausible that helix α0 in TIA-1-RRM3 directly enhances RNA binding and thus provides additional contacts for RNA recognition. Residues in helix α0 (YDEVV) could mediate such additional contacts. Figure 3B (box) shows that the side chains of Val194 and Leu189 pack against helix α2 in RRM3, while Asp192 and Glu193 are solvent exposed. However, Tyr191 is partially exposed and may thus mediate additional contacts with the RNA ligands, thereby contributing to RNA binding. Taken together our data indicate that the tandem RRM2,3 domain of TIA-1 represents two canonical β-sheet surfaces for RNA binding that are extended by the novel helix α0. High-resolution structural analysis of the RRM2,3/RNA complex will be required to unravel how both RRM domains cooperate in RNA recognition and how helix α0 enhances these interactions.
The NMR titrations of the three individual RRMs of TIA-1 with U9 RNA confirm previous reports that RRM1 does not bind RNA and that RRM2 and RRM3 mediate the specific RNA binding by TIA-1 (12,17). ITC measurements indicate the presence of two binding sites in fas_16 RNA for TIA-1-RRM2,3. The higher affinity site (KD = 0.35 nM) is 20-fold stronger than U9 (KD = 7 nM). However, the higher affinity is most likely due to avidity effects as RRM2,3 has comparable binding affinity to a shorter RNA derived from fas_16 (fas_10, 5′-UUCUCCUUUC-3′) or U9 RNA. This suggests, that TIA-1 does not discriminate between different poly-pyrimidine stretches and that binding affinity can be significantly increased by avidity effects, as seen with other protein–ligand interactions (24,56) and consistent with the presence of longer (>10 nt) pyrimidine-rich sequences in the RNA targets of TIA-1.
A number of non-canonical RRM domains with degenerate RNP1 and RNP2 motifs have been found to mediate protein–protein interactions rather than RNA recognitions. These non-canonical RRMs, also called U2AF-homology motifs (UHMs) (57), can bind to protein partners through a tryptophan-containing peptide motif (UHM-ligand motifs, ULM) (58–61) and often play roles in the regulation of alternative splicing (60–61). The distinct characteristics of UHMs compared to canonical RRM domains encompass atypical RNP-like motifs, an RXY/F motif in the last loop, and an acidic extension of the first helix. Notably, some of these features are observed for TIA-1 RRM1, i.e. the first RNP1 residue is aspartic acid instead of lysine residue in both RRM2 and RRM3 (Figure 1B). RRM1 harbors an RXI/L/V motif in a C-terminal loop that resembles the characteristic RXY/F motif of UHM domains. Although TIA-1-RRM1 is overall positively charged our NMR titrations with poly-uridine–RNA demonstrates that RRM1 only weakly interacts with RNA and mediates primarily non-specific interactions. Indeed, the binding of RRM1,2,3 to U15 RNA shows comparable affinities than RRM2,3 to U9 RNA, suggesting that RRM1 is dispensable for high affinity RNA binding. It is thus likely that a main function of RRM1 is to mediate protein–protein rather than RNA interactions. Consistent with this notion is the recent observation that RRM1 is dispensable for binding to an AU-rich RNA ligand (24). Some minor contribution to binding of poly-U RNA reported by the same authors may indeed reflect non-specific RRM1 interactions, consistent with our NMR titrations. A recent study by Kielkopf et al. concluded that RRM1 contributes to high affinity RNA binding to poly-uridine RNA (U20) but not to FAS RNA. In this study mutations of the RNP motifs in RRM1 decreased the affinity to U20 RNA (621). It is possible that the elimination of weak contacts of RRM1 with the RNA influences local concentration or avidity effects. This could also explain why the same authors did not observe a contribution of RRM1 in binding to a 31 nt FAS RNA ligand, which contains pyrimidine-stretches interspersed with purine bases and thus may not support avidity effects.
In recent years, it has been shown that domain arrangements and dynamics play an important role for molecular function and ligand binding of multi-domain proteins. Although many RRM–RNA complex structures have been reported, most involve individual RNA-binding domains bound to single-stranded RNAs (56,62–64. While these structures reveal details of the RNA recognition for individual domains, the role of multi-domain arrangements and dynamics is not analyzed. Weak domain interactions and dynamics can play important roles for ligand binding and the arrangements of multiple domains in solution might be different than observed by crystallographic studies (56,65). As TIA-1-RRM2 possesses the dominant RNA binding ability of TIA-1, RNA binding by RRM2,3 may utilize initial contacts by RRM2 to a specific RNA motif, while RRM3, connected with a 20-residue flexible linker might search for additional RNA motifs using a fly casting mechanism (Figure 8E).
Compared to other alternative splicing factors that bind close to either 3′ or 5′ splice sites (66–67), TIA-1 binding was only found downstream of exons to recruit U1 snRNP to the 5′ splice sites (12–13). For example, RNA binding of TIA-1 requires U1 snRNP to form a cooperative complex at a specific location in the pre-mRNA. Recent iCLIP data show that TIA-1 binds equally well to Py-tracts downstream of constitutive and alternative exons and its binding downstream of constitutive exons promotes efficient splicing of the corresponding intron, suggesting an importance of distal regulatory sites for the regulation of alternative splicing (26). Thus, more data are required to gain more details for how TIA-1 manages its recognition with various RNAs, especially with different RNA ligands downstream to the constitutive and alternative exons. Besides of binding to similar U-rich stretches, TIA-1 and TIAR recognize cytosine/uridine-rich (C-rich) elements according to SELEX analysis and gene array studies (4,68). Therefore, the higher affinity RNA binding site (KD1 ∼0.35 nM) in FAS intron 5 (fas_16) seen in our ITC analysis may represent such a cytosine and uridine-rich site and thus explain the specificity over poly-uridine RNA (U9). Future studies and structural analysis of protein–RNA complexes will help to unravel the sequence specificity of RNA recognition by TIA-1 and the molecular basis for the cooperative binding of TIA-1 and U1 snRNP to the pre-mRNA.
STRUCTURE COORDINATES AND NMR DATA
Atomic coordinates of TIA-1-RRM2,3 have been deposited in the RCSB Protein Data Bank with accession number 2MJN, while NMR chemical shift data and restraint files are available at the BMRB (accession number: 19735).
Accession Numbers
PDB: 2RNE, 3BS9 and 1X4G.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [1,2].
Acknowledgments
We thank Fatiha Kateb for help with NMR relaxation measurements and discussions, the ESRF for beamtime on BM29 and local contact Dr Louiza Zerrad for support and the Bavarian NMR Centre for NMR measurement time. We are grateful Clara Kielkopf for providing bacterial expression vectors for TIA-1.
FUNDING
Fellowship from the Postdoctoral Research Abroad Program sponsored by the National Science Council (NSC) of the R.O.C. (Taiwan) [to I.W.] ; fellowships from the Swedish Research Council (Vetenskapsrådet) and the European Molecular Biology Organization [EMBO, ALTF 276-2010 to J.H.]; Boehringer-Ingelheim funds for a Ph.D. fellowship [to P.K.A.J.]; Deutsche Forschungsgemeinschaft [SFB1035, GRK1721 to M.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Forch P., Puig O., Kedersha N., Martinez C., Granneman S., Seraphin B., Anderson P., Valcarcel J. The apoptosis-promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol. Cell. 2000;6:1089–1098. doi: 10.1016/s1097-2765(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 2.Zuccato E., Buratti E., Stuani C., Baralle F.E., Pagani F. An intronic polypyrimidine-rich element downstream of the donor site modulates cystic fibrosis transmembrane conductance regulator exon 9 alternative splicing. J. Biol. Chem. 2004;279:16980–16988. doi: 10.1074/jbc.M313439200. [DOI] [PubMed] [Google Scholar]
- 3.Singh N.N., Seo J., Ottesen E.W., Shishimorova M., Bhattacharya D., Singh R.N. TIA1 prevents skipping of a critical exon associated with spinal muscular atrophy. Mol. Cell. Biol. 2011;31:935–954. doi: 10.1128/MCB.00945-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez de Silanes I., Galban S., Martindale J.L., Yang X., Mazan-Mamczarz K., Indig F.E., Falco G., Zhan M., Gorospe M. Identification and functional outcome of mRNAs associated with RNA-binding protein TIA-1. Mol. Cell. Biol. 2005;25:9520–9531. doi: 10.1128/MCB.25.21.9520-9531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawai T., Lal A., Yang X., Galban S., Mazan-Mamczarz K., Gorospe M. Translational control of cytochrome c by RNA-binding proteins TIA-1 and HuR. Mol. Cell. Biol. 2006;26:3295–3307. doi: 10.1128/MCB.26.8.3295-3307.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piecyk M., Wax S., Beck A.R., Kedersha N., Gupta M., Maritim B., Chen S., Gueydan C., Kruys V., Streuli M., et al. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J. 2000;19:4154–4163. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Damgaard C.K., Lykke-Andersen J. Translational coregulation of 5’TOP mRNAs by TIA-1 and TIAR. Genes Dev. 2011;25:2057–2068. doi: 10.1101/gad.17355911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivanov P., Kedersha N., Anderson P. Stress puts TIA on TOP. Genes Dev. 2011;25:2119–2124. doi: 10.1101/gad.17838411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian Q., Taupin J., Elledge S., Robertson M., Anderson P. Fas-activated serine/threonine kinase (FAST) phosphorylates TIA-1 during Fas-mediated apoptosis. J. Exp. Med. 1995;182:865–874. doi: 10.1084/jem.182.3.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Izquierdo J.M., Majos N., Bonnal S., Martinez C., Castelo R., Guigo R., Bilbao D., Valcarcel J. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol. Cell. 2005;19:475–484. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 11.Cheng J., Zhou T., Liu C., Shapiro J.P., Brauer M.J., Kiefer M.C., Barr P.J., Mountz J.D. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263:1759–1762. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- 12.Forch P., Puig O., Martinez C., Seraphin B., Valcarcel J. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5’ splice sites. EMBO J. 2002;21:6882–6892. doi: 10.1093/emboj/cdf668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gesnel M.C., Theoleyre S., Del Gatto-Konczak F., Breathnach R. Cooperative binding of TIA-1 and U1 snRNP in K-SAM exon splicing activation. Biochem. Biophys. Res. Commun. 2007;358:1065–1070. doi: 10.1016/j.bbrc.2007.05.050. [DOI] [PubMed] [Google Scholar]
- 14.Del Gatto-Konczak F., Bourgeois C.F., Le Guiner C., Kister L., Gesnel M.C., Stevenin J., Breathnach R. The RNA-binding protein TIA-1 is a novel mammalian splicing regulator acting through intron sequences adjacent to a 5’ splice site. Mol. Cell. Biol. 2000;20:6287–6299. doi: 10.1128/mcb.20.17.6287-6299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aznarez I., Barash Y., Shai O., He D., Zielenski J., Tsui L.C., Parkinson J., Frey B.J., Rommens J.M., Blencowe B.J. A systematic analysis of intronic sequences downstream of 5′ splice sites reveals a widespread role for U-rich motifs and TIA1/TIAL1 proteins in alternative splicing regulation. Genome Res. 2008;18:1247–1258. doi: 10.1101/gr.073155.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Izquierdo J.M., Valcarcel J. Fas-activated serine/threonine kinase (FAST K) synergizes with TIA-1/TIAR proteins to regulate Fas alternative splicing. J. Biol. Chem. 2007;282:1539–1543. doi: 10.1074/jbc.C600198200. [DOI] [PubMed] [Google Scholar]
- 17.Dember L.M., Kim N.D., Liu K.Q., Anderson P. Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities. J. Biol. Chem. 1996;271:2783–2788. doi: 10.1074/jbc.271.5.2783. [DOI] [PubMed] [Google Scholar]
- 18.Forch P., Valcarcel J. Molecular mechanisms of gene expression regulation by the apoptosis-promoting protein TIA-1. Apoptosis. 2001;6:463–468. doi: 10.1023/a:1012441824719. [DOI] [PubMed] [Google Scholar]
- 19.Izquierdo J.M., Valcarcel J. Two isoforms of the T-cell intracellular antigen 1 (TIA-1) splicing factor display distinct splicing regulation activities. Control of TIA-1 isoform ratio by TIA-1-related protein. J. Biol. Chem. 2007;282:19410–19417. doi: 10.1074/jbc.M700688200. [DOI] [PubMed] [Google Scholar]
- 20.Reyes R., Alcalde J., Izquierdo J.M. Depletion of T-cell intracellular antigen proteins promotes cell proliferation. Genome Biol. 2009;10:R87. doi: 10.1186/gb-2009-10-8-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R., et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 22.Gouet P., Courcelle E., Stuart D.I., Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 23.Gouet P., Robert X., Courcelle E. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bauer W.J., Heath J., Jenkins J.L., Kielkopf C.L. Three RNA recognition motifs participate in RNA recognition and structural organization by the pro-apoptotic factor TIA-1. J. Mol. Biol. 2011;45:727–740. doi: 10.1016/j.jmb.2011.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cruz-Gallardo I., Aroca A., Persson C., Karlsson B.G., Diaz-Moreno I. RNA binding of T-cell intracellular antigen-1 (TIA-1) C-terminal RNA recognition motif is modified by pH conditions. J. Biol. Chem. 2013;288:25986–25994. doi: 10.1074/jbc.M113.489070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z., Kayikci M., Briese M., Zarnack K., Luscombe N.M., Rot G., Zupan B., Curk T., Ule J. iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLoS Biol. 2010;8:e1000530. doi: 10.1371/journal.pbio.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar A.O., Swenson M.C., Benning M.M., Kielkopf C.L. Structure of the central RNA recognition motif of human TIA-1 at 1.95A resolution. Biochem. Biophys. Res. Commun. 2008;367:813–819. doi: 10.1016/j.bbrc.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuwasako K., Takahashi M., Tochio N., Abe C., Tsuda K., Inoue M., Terada T., Shirouzu M., Kobayashi N., Kigawa T., et al. Solution structure of the second RNA recognition motif (RRM) domain of murine T cell intracellular antigen-1 (TIA-1) and its RNA recognition mode. Biochemistry. 2008;47:6437–6450. doi: 10.1021/bi7024723. [DOI] [PubMed] [Google Scholar]
- 29.Aroca A., Diaz-Quintana A., Diaz-Moreno I. A structural insight into the C-terminal RNA recognition motifs of T-cell intracellular antigen-1 protein. FEBS Lett. 2011;585:2958–2964. doi: 10.1016/j.febslet.2011.07.037. [DOI] [PubMed] [Google Scholar]
- 30.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 31.Goddard T.D., Kneller D.G. SPARKY. :3. [Google Scholar]
- 32.Johnson B.A., Blevins R.A. NMR view: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 33.Sattler M., Schleucher J., Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999;34:93–158. [Google Scholar]
- 34.Grzesiek S., Bax A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 35.Yamazaki T., Forman-Kay J.D., Kay L.E. Two-dimensional NMR experiments for correlating carbon-13.beta. and proton.delta./.epsilon. chemical shifts of aromatic residues in 13C-labeled proteins via scalar couplings. J. Am. Chem. Soc. 1993;115:11054–11055. [Google Scholar]
- 36.Tjandra N., Kuboniwa H., Ren H., Bax A. Rotational dynamics of calcium-free calmodulin studied by 15N-NMR relaxation measurements. Eur. J. Biochem. 1995;230:1014–1024. doi: 10.1111/j.1432-1033.1995.tb20650.x. [DOI] [PubMed] [Google Scholar]
- 37.Massi F., Johnson E., Wang C., Rance M., Palmer A.G., 3rd NMR R1 rho rotating-frame relaxation with weak radio frequency fields. J. Am. Chem. Soc. 2004;126:2247–2256. doi: 10.1021/ja038721w. [DOI] [PubMed] [Google Scholar]
- 38.Daragan V.A., Mayo K.H. Motional model analyses of protein and peptide dynamics using 13C and 15N NMR relaxation. Prog. Nucl. Magn. Reson. Spectrosc. 1997;31:63–105. [Google Scholar]
- 39.Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003;36:1277–1282. [Google Scholar]
- 40.Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992;25:495–503. [Google Scholar]
- 41.Jacques D.A., Guss J.M., Svergun D.I., Trewhella J. Publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution. Acta Crystallogr. D Biol. Crystallogr. 2012;68:620–626. doi: 10.1107/S0907444912012073. [DOI] [PubMed] [Google Scholar]
- 42.Guntert P. Automated structure determination from NMR spectra. Eur. Biophys. J. 2009;38:129–143. doi: 10.1007/s00249-008-0367-z. [DOI] [PubMed] [Google Scholar]
- 43.Shen Y., Delaglio F., Cornilescu G., Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nederveen A.J., Doreleijers J.F., Vranken W., Miller Z., Spronk C.A., Nabuurs S.B., Guntert P., Livny M., Markley J.L., Nilges M., et al. RECOORD: a recalculated coordinate database of 500 +proteins from the PDB using restraints from the BioMagResBank. Proteins. 2005;59:662–672. doi: 10.1002/prot.20408. [DOI] [PubMed] [Google Scholar]
- 45.Doreleijers J.F., Sousa da Silva A.W., Krieger E., Nabuurs S.B., Spronk C.A., Stevens T.J., Vranken W.F., Vriend G., Vuister G.W. CING: an integrated residue-based structure validation program suite. J. Biomol. NMR. 2012;54:267–283. doi: 10.1007/s10858-012-9669-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koradi R., Billeter M., Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 47.Walsh J.D., Meier K., Ishima R., Gronenborn A.M. NMR studies on domain diffusion and alignment in modular GB1 repeats. Biophys. J. 2010;99:2636–2646. doi: 10.1016/j.bpj.2010.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mulder F.A., Schipper D., Bott R., Boelens R. Altered flexibility in the substrate-binding site of related native and engineered high-alkaline Bacillus subtilisins. J. Mol. Biol. 1999;292:111–123. doi: 10.1006/jmbi.1999.3034. [DOI] [PubMed] [Google Scholar]
- 49.Consortium T.U. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2011;40:D71–75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schumann F.H., Riepl H., Maurer T., Gronwald W., Neidig K.P., Kalbitzer H.R. Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR. 2007;39:275–289. doi: 10.1007/s10858-007-9197-z. [DOI] [PubMed] [Google Scholar]
- 51.Garcia de la Torre J., Huertas M.L., Carrasco B. HYDRONMR: prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J. Magn. Reson. 2000;147:138–146. doi: 10.1006/jmre.2000.2170. [DOI] [PubMed] [Google Scholar]
- 52.Daubner G.M., Clery A., Allain F.H. RRM-RNA recognition: NMR or crystallography…and new findings. Curr. Opin. Struct. Biol. 2013;23:100–108. doi: 10.1016/j.sbi.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 53.Netter C., Weber G., Benecke H., Wahl M.C. Functional stabilization of an RNA recognition motif by a noncanonical N-terminal expansion. RNA. 2009;15:1305–1313. doi: 10.1261/rna.1359909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perez Canadillas J.M., Varani G. Recognition of GU-rich polyadenylation regulatory elements by human CstF-64 protein. EMBO J. 2003;22:2821–2830. doi: 10.1093/emboj/cdg259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacks A., Babon J., Kelly G., Manolaridis I., Cary P.D., Curry S., Conte M.R. Structure of the C-terminal domain of human La protein reveals a novel RNA recognition motif coupled to a helical nuclear retention element. Structure. 2003;11:833–843. doi: 10.1016/s0969-2126(03)00121-7. [DOI] [PubMed] [Google Scholar]
- 56.Mackereth C.D., Sattler M. Dynamics in multi-domain protein recognition of RNA. Curr. Opin. Struct. Biol. 2012;22:287–296. doi: 10.1016/j.sbi.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 57.Kielkopf C.L., Lucke S., Green M.R. U2AF homology motifs: protein recognition in the RRM world. Genes Dev. 2004;18:1513–1526. doi: 10.1101/gad.1206204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kielkopf C.L., Rodionova N.A., Green M.R., Burley S.K. A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell. 2001;106:595–605. doi: 10.1016/s0092-8674(01)00480-9. [DOI] [PubMed] [Google Scholar]
- 59.Selenko P., Gregorovic G., Sprangers R., Stier G., Rhani Z., Kramer A., Sattler M. Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/mBBP. Mol. Cell. 2003;11:965–976. doi: 10.1016/s1097-2765(03)00115-1. [DOI] [PubMed] [Google Scholar]
- 60.Corsini L., Bonnal S., Basquin J., Hothorn M., Scheffzek K., Valcarcel J., Sattler M. U2AF-homology motif interactions are required for alternative splicing regulation by SPF45. Nat. Struct. Mol. Biol. 2007;14:620–629. doi: 10.1038/nsmb1260. [DOI] [PubMed] [Google Scholar]
- 61.Corsini L., Hothorn M., Stier G., Rybin V., Scheffzek K., Gibson T.J., Sattler M. Dimerization and protein binding specificity of the U2AF homology motif of the splicing factor Puf60. J. Biol. Chem. 2009;284:630–639. doi: 10.1074/jbc.M805395200. [DOI] [PubMed] [Google Scholar]
- 62.Tsuda K., Someya T., Kuwasako K., Takahashi M., He F., Unzai S., Inoue M., Harada T., Watanabe S., Terada T. Structural basis for the dual RNA-recognition modes of human Tra2-beta RRM. Nucleic Acids Res. 2011;39:1538–1553. doi: 10.1093/nar/gkq854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Handa N., Nureki O., Kurimoto K., Kim I., Sakamoto H., Shimura Y., Muto Y., Yokoyama S. Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature. 1999;398:579–585. doi: 10.1038/19242. [DOI] [PubMed] [Google Scholar]
- 64.Lunde B.M., Moore C., Varani G. RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mackereth C.D., Madl T., Bonnal S., Simon B., Zanier K., Gasch A., Rybin V., Valcarcel J., Sattler M. Multi-domain conformational selection underlies pre-mRNA splicing regulation by U2AF. Nature. 2011;475:408–411. doi: 10.1038/nature10171. [DOI] [PubMed] [Google Scholar]
- 66.Ule J., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 67.Xue Y., Zhou Y., Wu T., Zhu T., Ji X., Kwon Y.S., Zhang C., Yeo G., Black D.L., Sun H., et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol. Cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim H.S., Kuwano Y., Zhan M., Pullmann R., Jr, Mazan-Mamczarz K., Li H., Kedersha N., Anderson P., Wilce M.C., Gorospe M., et al. Elucidation of a C-rich signature motif in target mRNAs of RNA-binding protein TIAR. Mol. Cell. Biol. 2007;27:6806–6817. doi: 10.1128/MCB.01036-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.