


Abstract
Histone H2A ubiquitination plays critical roles in transcriptional repression and deoxyribonucleic acid (DNA) damage response. More attention has been focused on ubiquitin E3 ligases of H2A, however, less is known about the negative regulators of H2A ubiquitination. Here we identified HSCARG as a new negative regulatory protein for H2A ubiquitination and found a possible link between regulator of H2A ubiquitination and cell cycle. Mechanistically, HSCARG interacts with polycomb repressive complex 1 (PRC1) and deubiquitinase USP7 and inhibits PRC1 ubiquitination in a USP7-dependent manner. As ubiquitination of PRC1 is critical for its E3 ligase activity toward H2A, HSCARG and USP7 are further shown to decrease the level of H2A ubiquitination. Moreover, we demonstrated that HSCARG is involved in DNA damage response through affecting the level of H2A ubiquitination and localization of RAP80 at lesion points. Knockout of HSCARG results in persistent activation of checkpoint signaling and leads to cell cycle arrest. This study unravels a novel mechanism for the regulation of H2A ubiquitination and elucidates how regulators of H2A ubiquitination affect cell cycle.
INTRODUCTION
H2A is the first protein to be identified as being ubiquitinated (1). It is estimated that 5–15% of H2A is ubiquitinated in mammalian cells. The functions of H2A ubiquitination were poorly understood until recent studies showing that ubiquitinated H2A is correlated with gene repression and deoxyribonucleic acid (DNA) damage repair (2–8). Several ubiquitin E3 ligases responsible for H2A have been identified, however, relatively less is known about negative regulators of H2A ubiquitination. The level of H2A ubiquitination varies at different stages of the cell cycle (4,5,9–18). H2A ubiquitination is correlated with cell cycle progression, and abnormality in either of the E3 ligases or deubiquitinases of H2A leads to a decreased rate of cell growth (2,16,17,19). However, the detailed mechanism linking regulators of H2A ubiquitination and cell cycle is still incompletely understood.
Polycomb repressive complex 1 (PRC1) is an ubiquitin E3 ligase of H2A ubiquitination (2). The core components of PRC1 are RING1, RING2 and BMI1, of which RING2 is the catalytic protein. The E3 ligase activity of PRC1 is regulated at multiple levels, with the self-ubiquitination of RING2 being critical for its catalytic activity (20,21). The other components of PRC1 are also important for its catalytic activity, RING1 and BMI1 can strongly stimulate the E3 ligase activity of RING2 but the mechanism is still unclear (2,3,19). Recent studies show that USP7 can regulate RING2 ubiquitination, however, whether USP7 affects H2A ubiquitination remains unclear yet.
DNA damage in cells is readily induced by environmental agents or is generated spontaneously during DNA metabolism. It is estimated that each cell develops up to 105 spontaneous DNA lesions per day (22). In response to DNA damage, cells have evolved a complicated mechanism to survive and ensure accurate transmission of the genome. DNA double strand breaks (DSBs) are the most dangerous of all insults to cells. When damages occur, a cascade reaction mediated by ataxia telangiectasia mutated (ATM) or ataxia telangiectasia and Rad3-related (ATR) is activated and phosphorylates H2AX (also denoted as γH2AX) around the damage points (23,24). This is followed by H2A ubiquitination catalyzed by various E3 ligases (4,5,15). The ubiquitin chains of H2A then act as docking sites for repair proteins such as RAP80, Abraxas, BRCA1 and 53BP1 translocating to the damaged sites (14,25,26). Meanwhile, ATM/ATR activates the checkpoint signaling and halts the cell cycle progression until the damage points are repaired (27–30). If the damage is too severe to be repaired, the cell will undergo apoptosis (31).
HSCARG (also known as NmrA-like family domain containing 1, NMRAL1) is a recently characterized protein belonging to the short-chain dehydrogenase family but without dehydrogenase activity (32). To elucidate the functions of HSCARG in cells, we used a yeast two-hybrid screen. We found that HSCARG interacts with PRC1. HSCARG interacts with and relies on USP7 to inhibit PRC1 ubiquitination, which further decreases the level of H2A ubiquitination. In addition, we demonstrated that HSCARG is involved in the DNA damage response and that knockout of HSCARG activates the signaling of cell cycle checkpoint and results in an obvious reduction in cell growth rate.
MATERIALS AND METHODS
Antibodies and reagents
Monoclonal anti-Flag (F3165), anti-HA (H9658) and IgG (M5284) antibodies were purchased from Sigma (MO, USA); anti-Myc (M047–3), anti-histidine (D291–3) and anti-β-actin (PM053) were from MBL (Japan); anti-H2A (39209) was from Active Motif (CA, USA). The polyclonal antibodies anti-p21 (sc-397), anti-USP7 (sc-30164) and anti-USP11 (sc-134928) were from Santa Cruz Biotechnology (TX, USA); anti-γH2AX (05–636) was from Millipore (MA, USA); anti-CHK2 (BS1526), anti-pCHK2 (BS4043) and anti-TFIID (BS2262) were from Bioworld (MN, USA); anti-RING1 (AP14560a) was from Abgent (CA, USA); anti-RAP80 (3746) was from Epitomics (CA, USA); and anti-HSCARG was generated against purified recombinant HSCARG. Protein G was purchased from GE Healthcare (Shanghai, China), the Ni-NTA agarose was from Qiagen (Germany) and the protease inhibitor was from Calbiochem (MA, USA).
Plasmids and shRNA preparation
The complementary DNAs (cDNAs) of RING1, RING2 and BMI1 were kindly provided by Dr Hengbin Wang at University of Alabama at Birmingham. HSCARG, RING1, RING2 and BMI1 were cloned into the vector pRK-HA or pRK-Flag respectively. H2A was cloned into pRK-HA or pRK-Flag and H2B into pRK-HA. FLAG-USP7 was a kind gift from Dr Goedele Maertens at Cancer Research UK. HA-RAP80 was a kind gift from Dr Xiaochun Yu at the University of Michigan Medical School. Flag-USP11 and USP11 shRNA were provided by Drs Xiaojie Tan and Jianhua Yang at Baylor College of Medicine. The RNA interference (RNAi) sequence targeting HSCARG, 5′-CCACCTTCATCGTGACCAATT, and the control sequence, 5′-ACGTGACACGTTCGGAGAATT, were inserted into the GV248 vector. The siRNA targeting USP7 was purchased from Santa Cruz (sc-41521). The shRNA targeting USP7 was from OriGene (TR308454). All the plasmids were verified by DNA sequencing.
Cell culture and transfection
HCT116, HEK293T and HeLa cells were grown in Iscove's modified Dulbecco's medium (Invitrogen, CA, USA), supplemented with 10% fetal calf serum (Hyclone) at 37°C in 5% CO2. Transfections were performed using Mega Tran1.0 (Origene) according to the manufacturer's instructions.
Co-immunoprecipitation (co-IP) and western blot
co-IP and western blot analyses were performed following procedures described previously (33). Briefly, HEK293T cells were transfected with the indicated plasmids. At 48 h after transfection, cells were washed with phosphate buffered saline (PBS) buffer and lysed in modified Radio-Immunoprecipitation Assay (RIPA) buffer (50-mM Tris-HCl, pH 7.4, 150-mM NaCl, 1-mM ethylenediaminetetraacetic acid (EDTA) and protease inhibitor) and sonicated. After pre-clearing with protein-G-Sepharose beads, the supernatant was incubated with antibodies or control IgG at 4°C overnight, followed by incubation with 50-μl protein G Sepharose for 4 h. The samples were denatured and loaded on a sodium dodecyl sulphate-polyacrylamide gel electrophoresis(SDS-PAGE) gel and transferred to a nitrocellulose membrane (GE Healthcare). Membranes were probed with primary and secondary antibodies. The protein signals were detected and quantified using Odyssey Infrared Imaging System and software Odyssey V3.0 (LI-COR Biosciences, NE, USA).
Denaturing immunoprecipitation analysis
To detect the effect of HSCARG on H2A ubiquitination, denaturing immunoprecipitation analysis was performed. HEK293T cells were transfected with HA-H2A and His-ubiquitin, with or without Flag-HSCARG. At 48 h after transfection, cells were washed with PBS buffer and lysed in 400-μl SDS lysis buffer (10% SDS in PBS). The lysates were heated to 95°C and vortexed vigorously more than three times. Subsequently, 800-μl-modified RIPA lysis buffer (described above) was added to the lysates, which were then cooled on ice for 1 h and centrifuged at 18 000 g for 30 min at 4°C. The supernatant was then subjected to immunoprecipitation with anti-HA and western blot with the anti-His antibody.
His-ubiquitin pull-down analysis
HCT116 cells were transfected with His-ubiquitin, together with or without Flag-HSCARG. And then the effect of HSCARG on H2A ubiquitination was examined by His-ubiquitin pull-down analysis following the method described previously (34).
Cellular fractionation assay
Cellular fractionation assay was performed following the procedures described previously (35,36). Briefly, cells were washed with PBS buffer and lysed in buffer A (10-mM HEPES pH7.9, 10-mM KCl, 1.5-mM MgCl2, 0.34-M sucrose, 10% glycerol, 1-mM dithiothreitol, 0.1% Triton X-100 and protease inhibitor cocktail) on ice for 8 min. The supernatant (fraction S1, cytosol) was collected. Cell pellets were washed by buffer A twice and lysed in buffer B (3-mM EDTA, 0.2-mM ethylene glycol tetraacetic acid (EGTA), 1-mM dithiothreitol and protease inhibitor cocktail) on ice for 30 min. The insoluble fractions (fraction chr, chromatin) and soluble (fraction S2, nucleoplasm) fractions were separated by centrifugation (5 min, 1700 g, 4°C).
IR treatment
Ion radiation was delivered by the X-ray generator (RS 2000; 160 kV; 25 mA; dose rate, 1.43 Gy/min). Cells were treated with IR at the dosage of 10 Gy and then incubated for 1 h before harvesting.
Acid chromatin fractionation
Acid chromatin fractionation was carried out following the procedures described (37). Briefly, cells were washed with PBS buffer and lysed in modified RIPA buffer described above. Cell pellets were washed by distilled water twice and incubated with 0.2-M HCl for 10 min on ice. The soluble fraction was then neutralized with 1-M Tris-HCl pH 8.0 for western blot analysis.
Immunofluorescence
The changes in the subcellular localization of RAP80 under different treatments were investigated in HeLa cells by using immunofluorescence. HeLa cells were transfected with or without Flag-HSCARG, 23 h later, cells were either treated with IR at the dosage of 10 Gy or were not treated with IR and then incubated for 1 h. Immunofluorescence was then carried out following procedures described previously (38). Images were visualized under a confocal laser scanning microscope (Zeiss LSM-710 NLO & DuoScan, Germany) using a 40× objective lens. The number and intensity of IR-induced nuclear foci were quantified using the software Imaris 7.6 (Bitplane, UK). γH2AX was used as a DNA damage marker and indicated the lesion points. Nuclear DNA was stained with 1-μg/ml 4,6-diamidino-2-phenylindole (DAPI).
Construction of HeLa cell-based HSCARG−/− cell line
HeLa cell-based HSCARG−/− cell line was constructed by TALEN technology following the manufacture's instruction (SIDANSAI Biotechnology CO., LTD). Briefly, the sequences target for HSCARG (NL1: GGTGGACAAGAAACT, NR1: GGCTCACCTGTGCCT) was inserted into plasmids pTALEN-L and pTALEN-R, respectively, and verified by DNA sequencing. The two plasmids were co-transfected into HeLa cells and positive clones were selected by puromycin. The knockout effect of HSCARG was confirmed by western blot analysis.
Construction of HCT116 cell-based HSCARG−/− cell line
The approach for generating targeted cells with adeno-associated virus (AAV) was performed as described (39). Briefly, vector arms were created by PCR from normal human DNA using HiFi Taq (Invitrogen) and validated by sequencing prior to virus production and infection. The targeting AAV viruses were packaged in HEK293T cells by transfecting equal amounts of the targeting vector, pHelper and pRC plasmids. Viruses were harvested 72 h after transfection. The parental HCT116 cells were infected with indicated rAAV viruses. Stable G418-resistant clones were selected in the presence of 0.4 mg/ml for HCT116 and then screened for PCR screening as reported. After the first allele was targeted, the neomycin resistance gene was excised by Cre-recombinase and the targeted clones were retargeted to obtain homozygous knock-out cells.
Clonogenic survival assay
Cells were seeded at low density (about 300 each dish) in 20-mm dish in triplicates. Twenty-four hour later, cells were treated with different dosages of IR (2, 4 and 6 Gy) and incubated for 12 days. Finally cells were stained with trypan blue and counted. The number of survived cells was normalized to non-irradiated wild-type cells.
Cell growth curve
Cells transfected with indicated plasmids were seeded onto a 24-well plate at the initial cell densities of 5000 per milliliter. The number of living cells was counted each day for 7 days, and three wells were counted per day.
FACS analysis
Wild-type and HSCARG−/− HCT116 cells were seeded onto 35-mm dishes. Twenty-four hour later, cells were trypsinized into a single cell suspension and fixed with 70% ethanol, and then stained with a solution containing 1% fetal bovine serum (FBS), 50-μg/ml propidium iodide and 25-μg/ml RNase A. Fluorescence-activated cell sorting (FACS) analysis was performed using a FACS Calibur cytometer, and data were analyzed using the CellQuest Pro software (BD Biosciences).
Ribonucleic acid (RNA) isolation and RT-PCR
To detect the changes in the messenger RNA (mRNA) levels of related proteins in HSCARG−/− HCT116 cells, total RNA populations were extracted from the wild-type and HSCARG−/− HCT116 cells by using TRIzol Reagent (Invitrogen). The cDNA was amplified by reverse transcriptase coupled PCR (RT-PCR) (Promega) following the manufacturer's instructions. The primers used are shown in Supplementary Table S2. Quantitative RT-PCR was performed using SsoFast™ EvaGreen® Supermix (Bio-Rad, CA, USA). For analysis, the Ct values were normalized against β-actin and analyzed using the ΔΔCt method
RNA-seq analysis
RNA-seq analysis was performed by BGI. Briefly, the total RNA from wild-type or HSCARG−/− HCT116 cells was extracted, and then the mRNA was fragmented into short (∼200-bp) fragments, and first-strand cDNA was synthesized by random hexamer primers using mRNA fragments as templates; this was followed by second-strand cDNA synthesis. Finally, all the fragments were subjected to Illumina Solexa ultrasequencing and mapped to different genes. Further analysis was carried out to examine the relative expression level of each gene.
RESULTS
HSCARG interacts with RING1 and inhibits RING1 ubiquitination
To define the functions of HSCARG, a yeast two-hybrid screen was used with full-length HSACRG as the bait. RING1, a core component of PRC1 that mediates the monoubiquitination of histone H2A (2,40,41), was identified as one of the target proteins that may associate with HSCARG. The interaction between HSCARG and RING1 was confirmed by both ectopic and endogenous co-IP (Supplementary Figure S1A and Figure 1A). Next, we examined the distribution of endogenous HSCARG and RING1 in HeLa cells by immunofluorescence analysis. The result showed that HSCARG co-localized with RING1 mainly in the nucleus, which further validates that HSCARG interacts with RING1 (Figure 1B).
Figure 1.
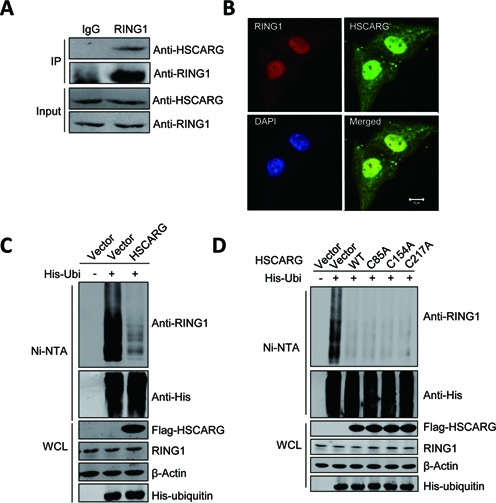
HSCARG interacts with RING1 and inhibits RING1 ubiquitination. (A) Endogenous interaction of HSCARG with RING1. HEK293T cells were harvested 48 h after seeding, and 5% of the cell lysates were analyzed directly (input). The remaining cell lysates were immunoprecipitated with anti-RING1 antibody or control rabbit IgG, followed by western blot with anti-RING1 and anti-HSCARG antibodies. (B) Co-localization of RING1 and HSCARG in HeLa cells. Subcellular localization of endogenously expressed RING1 and HSCARG was visualized by immunofluorescence microscopy using anti-RING1 antibody (red), anti-HSCARG antibody (green) and DAPI (blue). Scale bar, 10 μm. (C) and (D) HSCARG inhibits endogenous RING1 ubiquitination. HEK293T cells were transfected with indicated plasmids. At 48 h after transfection, cells were lysed with denaturing buffer and incubated with Ni-NTA beads, and the ubiquitinated proteins were purified and subjected to western blot with the indicated antibodies.
We next assessed whether HSCARG interacts with the other core components of the PRC1, RING2 or BMI1, two proteins that interact with RING1 (41). As expected, co-IP showed that HSCARG also interacted with RING2 and BMI1 (Supplementary Figure S1A). Taken together, these results indicate that HSCARG interacts with the core components of the PRC1, RING1, RING2 and BMI1.
As HSCARG interacts with RING1, we examined whether HSCARG affects the expression of RING1, and no visible effect was observed (Supplementary Figure S1C). A previous study shows that RING1 could be modified by ubiquitin chains (35), we next tested whether HSCARG affects ubiquitination of RING1. His-ubiquitin pull-down analysis showed that HSCARG clearly reduced the ubiquitin chains of endogenous RING1 (Figure 1C). This result promoted us to investigate whether HSCARG is a deubiquitinase of RING1. As cysteine is the vital residue for the activity of deubiquitinase (42–44), we mutated all the three cysteine residues of HSCARG and detected whether these mutants still work. As shown in Figure 1D, all the HSCARG mutants are as efficient as wild type in inhibition of RING1 ubiquitination. Further sequence analysis revealed that there is no conserved deubiquitinase domain in HSCARG. These results exclude the possibility that HSCARG is a deubiquitinase.
Our previous results show that HSCARG is a sensor of reduced nicotinamide adenine dinucleotide phosphate (NADPH) and residues Arg37, Tyr81, Lys133 are critical for its NADPH binding activity (45). Here we mutated these residues and tested if NADPH binding ability of HSCARG is critical for its function in inhibiting RING1 ubiquitination. The result showed that all the mutants still worked as efficiently as wild-type HSCARG (Supplementary Figure S1B). Taken together, these results indicate that HSCARG interacts with RING1 and inhibits RING1 ubiquitination, which is independent of its NADPH binding activity.
HSCARG depends on USP7 in inhibiting PRC1 ubiquitination
Recent reports show that USP7 could remove ubiquitin chains from PRC1 (35,46), we next investigated if there is a correlation between HSCARG and USP7. As shown in Supplementary Figure S2A, HSCARG interacted with USP7 and USP7 interacted with RING1 as well. Besides, HSCARG and RING1 could be precipitated by USP7 simultaneously. To further investigate the relation among HSCARG, RING1 and USP7, we co-transfected cells with USP7, RING1 and HSCARG and used one protein as bait to detect the other two proteins. The results showed that the three proteins could interact with each other (Figure 2A). These results indicate that USP7, RING1 and HSCARG form a complex.
Figure 2.
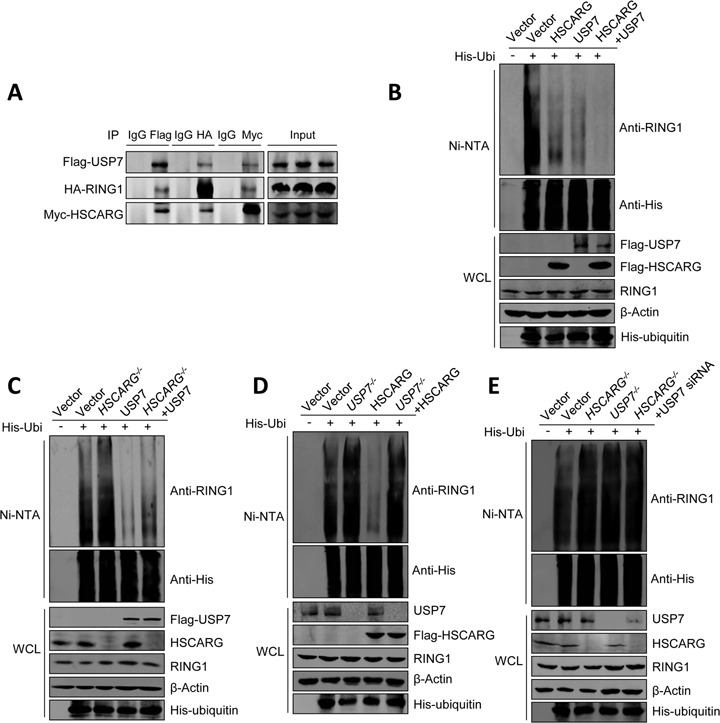
HSCARG depends on USP7 in inhibiting RING1 ubiquitination. (A) HSCARG, RING1 and USP7 form a complex. HEK293T cells were transiently transfected with Flag-USP7, HA-RING1 and Myc-HSCARG. Forty-eight hour later, Co-IP analysis was performed with anti-Flag/HA/Myc antibodies or control mouse IgG, followed by western blot with anti-Flag, anti-HA and anti-Myc antibodies. (B)–(E) HSCARG depends on USP7 in inhibiting RING1 ubiquitination. The level of endogenous RING1 ubiquitination was monitored by His-ubiquitin pull-down analysis in HEK293T cells (B) or HSCARG−/−, USP7−/− and wild-type HCT116 cells transfected with indicated plasmids (C)–(E).
Next, we examined the effects of USP7 and HSCARG on ubiquitination of RING1. His-ubiquitin pull-down analysis showed that HSCARG and USP7 inhibited RING1 ubiquitination in a synergy manner (Figure 2B). In addition, denaturing immunoprecipitation and His-ubiquitin pull-down assays showed that HSCARG functioned cooperatively with USP7 in inhibiting the ubiquitination of RING2 and BMI1 (Supplementary Figure S2B and C).
To further elucidate the relation between HSCARG and USP7, we constructed two HSCARG−/− HCT116 cell lines by using the Cre/loxP system to insert an additional sequence into the fourth exon and added further stop codons to disrupt HSCARG translation (Supplementary Figure S3A). Knockout of HSCARG was confirmed by both RT-PCR (Supplementary Figure S3B) and western blot analyses (Supplementary Figure S3C). A USP7−/− HCT116 cell line from Dr Bert Vogelstein was also used to study the effect of USP7 on HSCARG-inhibited RING1 ubiquitination.
As expected, deletion of HSCARG resulted in an increase in RING1 ubiquitination and attenuated the inhibition of RING1 ubiquitination by USP7 (Figure 2C), while in USP7−/− cells, the activity of HSCARG was completely abolished (Figure 2D). This result indicates that HSCARG relies on USP7 in inhibiting RING1 ubiquitination. To validate this observation, we detected the ubiquitination level of RING1 when both HSCARG and USP7 were depleted. The results showed that, compare to cells with individually depleted HSCARG or USP7, disruption of both HSCARG and USP7 resulted in no more increased RING1 ubiquitination (Figure 2E).
Collectively, these results indicate that HSCARG depends on USP7 in inhibiting RING1 ubiquitination.
HSCARG inhibits PRC1-mediated H2A ubiquitination
The PRC1 is an important E3 ubiquitin-protein ligase that mediates monoubiquitination at Lys-119 of histone H2A and the ubiquitination of PRC1 itself is critical for its catalytic activity (2,20). The result that HSCARG inhibits PRC1 ubiquitination promoted us to determine whether HSCARG also affects the ubiquitination of H2A. HEK293T cells were transfected with HA-H2A and His-ubiquitin together with or without Flag-HSCARG, and denaturing immunoprecipitation assay was performed. As expected, HSCARG strongly reduced H2A ubiquitination (Figure 3A). To further confirm this observation, His-ubiquitin pull-down assay was performed to detect the effect of HSCARG on endogenous H2A ubiquitination. Consistent with the exogenous results, HSCARG strongly reduced the endogenous H2A ubiquitination and all the mutants of HSCARG functioned similarly to the wild-type HSCARG (Figure 3B and Supplementary Figure S1D and E). These results indicate that HSCARG inhibits H2A ubiquitination independent of its NADPH binding activity.
Figure 3.
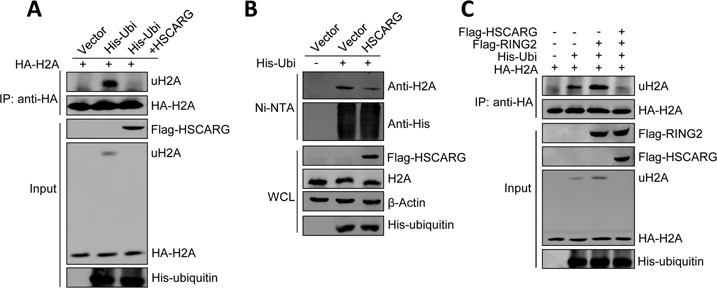
HSCARG inhibits PRC1-mediated H2A ubiquitination. (A) HSCARG suppresses H2A ubiquitination. HEK293T cells were transfected with HA-H2A, His-ubiquitin and Flag-HSCARG as indicated. Forty-eight hour later, cells were harvested and lysed in denaturing buffer, and H2A conjugates were isolated by immunoprecipitation using the anti-HA antibody and protein G. The bound complexes were analyzed by western blot with anti-His and anti-HA antibodies. (B) HSCARG inhibits endogenous H2A ubiquitination. HEK293T cells were transfected with His-ubiquitin and Flag-HSCARG as indicated. Forty-eight hour later, His-ubiquitin pull-down analysis was performed using indicated antibody. (C) HSCARG reduces RING2-catalyzed H2A ubiquitination. HEK293T cells were transfected with HA-H2A, His-ubiquitin, Flag-HSCARG and Flag-RING2 as indicated. Forty-eight hour later, the levels of ubiquitinated H2A were monitored by denaturing immunoprecipitation analysis as previously described followed by western blot with indicated antibodies.
Considering RING2 is the catalytic protein of PRC1, we then performed a denaturing immunoprecipitation assay to investigate whether HSCARG affects the catalytic activity of RING2. As expected, RING2 increased the level of ubiquitinated H2A, which was obviously decreased by HSCARG overexpression (Figure 3C). These results indicate that HSCARG inhibits PRC1-mediated H2A ubiquitination.
HSCARG depends on USP7 in inhibiting H2A ubiquitination
The above results indicate that proteins that affect PRC1 ubiquitination will also affect H2A ubiquitination, we therefore examined whether USP7 could also inhibit H2A ubiquitination. Indeed, denaturing immunoprecipitation and His-ubiquitin pull-down analysis showed that USP7 inhibited both exogenous and endogenous H2A ubiquitination (Figure 4A and B). We next ascertained how HSCARG and USP7 work in inhibiting H2A ubiquitination. As shown in Figure 4C, these two proteins inhibited H2A ubiquitination in a cooperative manner, as co-expression of HSCARG and USP7 almost completely inhibited H2A ubiquitination.
Figure 4.
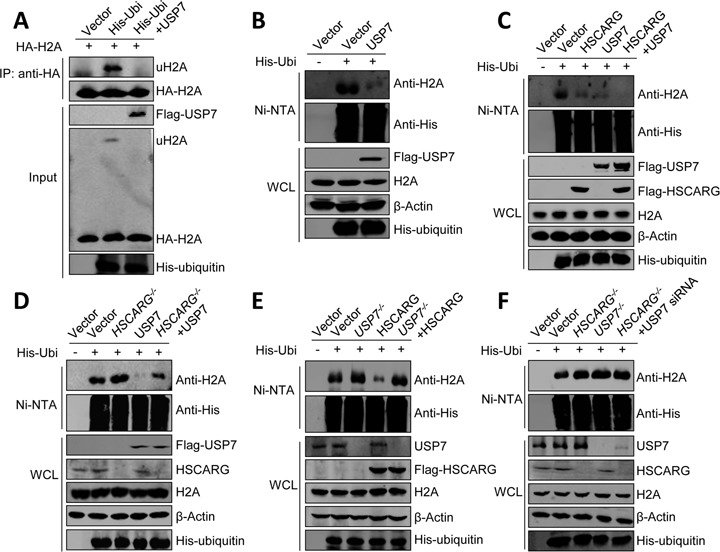
HSCARG depends on USP7 in inhibiting H2A ubiquitination. (A) USP7 inhibits H2A ubiquitination. HEK293T cells were transfected with HA-H2A, His-ubiquitin and Flag-USP7 as indicated. Forty-eight hour later, denaturing immunoprecipitation was performed using the anti-HA antibody and protein G. The bound complexes were analyzed by western blot with anti-His and anti-HA antibodies. (B) USP7 inhibits endogenous H2A ubiquitination. HEK293T cells transfected with His-ubiquitin and Flag-USP7 were subjected to His-ubiquitin pull-down assay using indicated antibody. (C)–(F) HSCARG depends on USP7 in inhibiting H2A ubiquitination. The level of endogenous H2A ubiquitination was monitored by His-ubiquitin pull-down analysis in HEK293T cells (C) or HSCARG−/−, USP7−/− and wild-type HCT116 cells transfected with indicated plasmids (D)–(F).
Next, we further investigated the relationship between HSCARG and USP7 in inhibiting H2A ubiquitination by using HSCARG−/− and USP7−/− cells. The results showed that the deubiquitinase activity of USP7 was obviously dampened but still remained to some extent in HSCARG−/− cells, while the activity of HSCARG was completely lost in USP7−/− cells (Figure 4D and E). Furthermore, deletion of both HSCARG and USP7 showed no obvious change in H2A ubiquitination compared to cells with individually depleted HSCARG or USP7. These results indicate that HSCARG depends on USP7 in inhibiting H2A ubiquitination.
HSCARG is involved in DNA damage response
H2A ubiquitination plays various roles in cellular processes such as transcriptional regulation and DNA damage repair (6–8,47–50). Since HSCARG inhibits H2A ubiquitination, we tested whether HSCARG is involved in ionizing radiation (IR)-induced DNA damage response as well. We first examined whether HSCARG inhibited DNA damage-induced H2A ubiquitination. HCT116 cells transfected with His-ubiquitin together with or without Flag-HSCARG were treated with IR (10 Gy) and the levels of H2A ubiquitination, a key marker of DNA damage, were monitored. The results showed that HSCARG did inhibit IR-induced H2A ubiquitination (Figure 5A).
Figure 5.
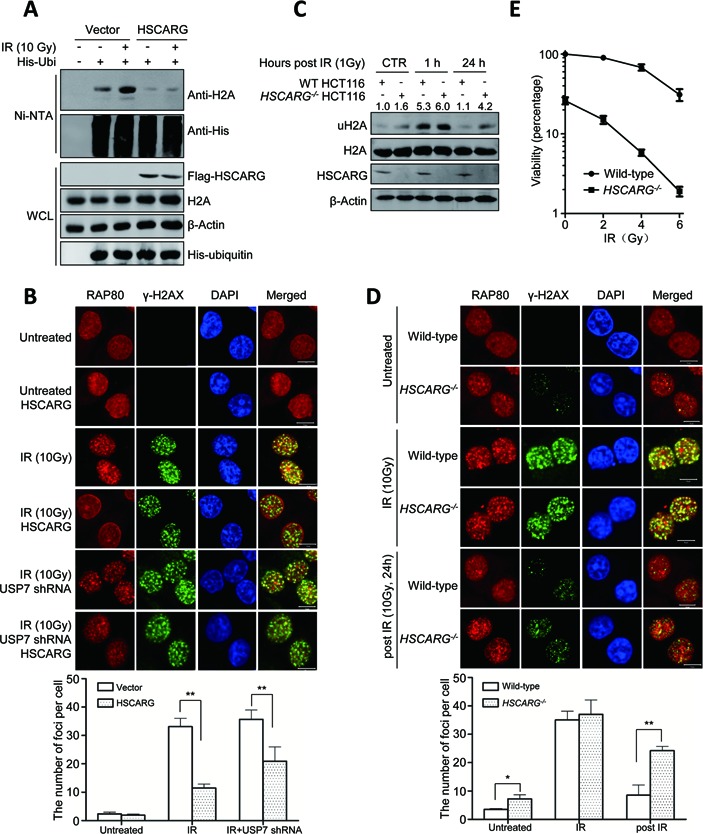
HSCARG affects DNA damage response. (A) HSCARG inhibits IR-induced H2A ubiquitination. HCT116 cells were treated with IR (10 Gy) and incubated for 1 h and then the level of H2A ubiquitination was detected by His-ubiquitin pull-down analysis. (B) HSCARG affects subcellular location of endogenous RAP80 in response to DNA damage. HeLa cells were transfected with or without Flag-HSCARG. For the USP7-depleted group, to get efficient USP7-knockdown cells, HeLa cells were transfected with USP7 shRNA and selected with puromycin before used; the knockdown effect was examined in Supplementary Figure S4A. At 24 h after transfection, cells were treated with IR (10 Gy), and 1 h later, cells were stained using anti-RAP80 antibody (red), anti-γ-H2AX antibody (green) and DAPI (blue). The average number of RAP80 foci per cell was quantified in at least 300 cells per sample. Error bars represent the SD of five independent experiments. P value was determined by Student's t-test. **indicates P < 0.01. Scale bar, 10 μm. (C) HSCARG deletion increases the persistence of H2A ubiquitination in response to IR. Both wild-type and HSCARG−/− HCT116 cells were treated with or without IR (1 Gy), cells were harvested at indicated time and the endogenous level of H2A ubiquitination was detected and quantified. The levels of uH2A were normalized against the total amount of uH2A and H2A. (D) RAP80 was trapped in HSCARG−/− HeLa cells. The subcellular location of endogenous RAP80 was detected in both wild-type and HSCARG−/− HeLa cells with indicated treatments. The average number of RAP80 foci per cell was quantified in at least 300 cells per sample. Error bars represent the SD of five independent experiments. P value was determined by Student's t-test. **indicates P < 0.01. *indicates P < 0.05. Scale bar, 10 μm. (E) HSCARG−/− cells are more sensitive to IR. Clonogenic survival assay was performed (see the Materials and Methods section). Data are from three independent experiments.
The ubiquitin chains of H2A provide docking sites for the accumulation of repair proteins at the damage lesions. RAP80 is a linker between H2A ubiquitination and repair proteins, it gathers at the lesion points and dissociates after the DNA damage repair (13,14,25). We performed immunofluorescence to determine whether HSCARG affected the localization of endogenous RAP80 at the lesion points. HeLa cells were transfected with or without HSCARG, and then treated with or without IR. Subsequently, γ-H2AX and the subcellular location of RAP80 were examined. In IR-treated cells, RAP80 gathered at the lesion points and co-localized with γ-H2AX, whereas in cells expressing HSCARG, the foci of RAP80 was decreased at the lesion points (Figure 5B). As HSCARG depends on USP7 in inhibiting H2A ubiquitination, we then assessed if depletion of USP7 rescued the decreased RAP80 foci caused by ectopic HSCARG. As shown in Figure 5B, knockdown of USP7 partially rescued RAP80 foci, which further supports that inhibition of H2A ubiquitination by HSCARG depends on USP7. These results indicate that HSCARG participates in the DNA damage response.
To further elucidate the function of HSCARG in DNA damage response, we detected the endogenous H2A ubiquitination in both wild-type and HSCARG−/- HCT116 cells at normal, IR-treated and post-IR-treated conditions. The results showed that under normal condition, the level of ubiquitinated H2A in HSCARG−/− cells was a little higher than that of wild-type cells, although they were relative lower in both cell lines when compared to the IR-treated cells. In cells treated with IR, the level of ubiquitinated H2A increased significantly in both cell lines, and at 24 h after IR treatment, in comparison to wild-type HCT116 cells, HSCARG−/− cells showed an obvious higher level of ubiquitinated H2A (Figure 5C).
The aforementioned results promoted us to investigate the localization of RAP80 in wild-type and HSCARG−/− cells at normal, IR-treated and post-IR-treated conditions. For immunofluorescence assay, we constructed HeLa-based HSCARG−/− cell by TALEN technology (Supplementary Figure S4B). The immunofluorescence results showed that, under normal condition, the number of RAP80 foci in HSCARG−/− cells was a little higher than that of wild-type cells. While in cells with IR treatment, the number of RAP80 foci increased significantly in both cell lines, and at 24 h after IR treatment, HSCARG−/− cells showed more foci in comparison to wild-type HCT116 cells (Figure 5D). These data are consistent with the observations of H2A ubiquitination. Altogether, these results indicate that HSCARG participates in the DNA damage response and affects the localization of repair protein RAP80 at the lesion points.
The involvement of HSCARG in DNA damage response drove us to further determine whether HSCARG affects the cell survival after IR treatment by clonogenic survival assay. Both wild-type and HSCARG−/- HCT116 cells were treated with different dosages of IR and the survival cells were counted. The result showed that the survival ratio of both wild-type and HSCARG−/- HCT116 cells decreased with the dosages of IR increased; moreover, cells without HSCARG were more sensitive to IR (Figure 5E). Collectively, these results indicate that HSCARG is involved in DNA damage response.
Deletion of HSCARG results in persistent activation of checkpoint signaling and reduces cell proliferation
To further elucidate the function of HSCARG in DNA damage response, the transcriptional differences between wild-type and HSCARG−/− HCT116 cells were compared by RNA-Seq analysis. Consistent with above results, the levels of several transcripts that are involved in DNA damage response changed significantly in HSCARG−/- cells where endogenous DNA damage may occur (Supplementary Table S1). Among these proteins, we noticed that the level of CDC25A/B/C decreased in HSCARG−/− cells (Supplementary Table S1), which is most likely caused by CHK2 phosphorylation. Indeed, western blot analysis showed that the Thr62 of CHK2 was phosphorylated in HSCARG−/− cells, whereas no signal was detected in wild-type cells (Figure 6A). Furthermore, we evaluated the phosphorylation of CHK2 in both wild-type and HSCARG−/− cells at IR-treated and post-IR-treated conditions. In response to IR treatment, the level of p-CHK2 increased obviously, but was a little bit higher in HSCARG−/− cells than that in wild-type cells. At a later stage after IR treatment (24 h), p-CHK2 reduced in both cells, but decreased slowly in HSCARG−/− cells (Figure 6A).
Figure 6.
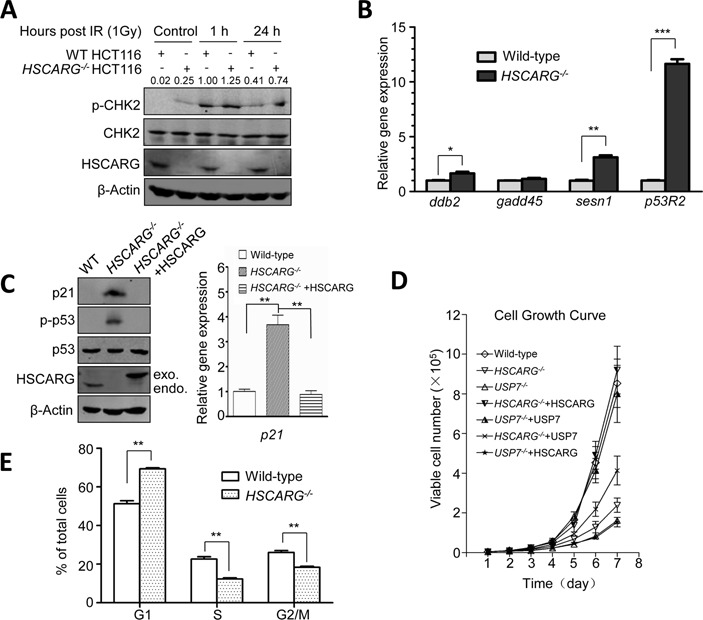
Deletion of HSCARG results in persistent activation of checkpoint signaling and reduces cell proliferation. (A) The phosphorylation level of CHK2 is increased in HSCARG−/− cells. Wild-type and HSCARG−/− HCT116 cells were either treated with IR (1 Gy) or were not treated with IR, and the levels of CHK2 and p-CHK2 were detected at indicated time and quantified. The levels of p-CHK2 were normalized against the total amount of p-CHK2 and CHK2. (B) The mRNA levels of ddb2, sesn1 and p53R2 are increased in HSCARG−/− cells. The total RNA populations of wild-type and HSCARG−/− HCT116 cells were extracted, after which qRT-PCR was performed to detect the mRNA levels of ddb2, gadd45, sesn1 and p53Rs. Values were reported as mean ± SD; P value was determined by Student's t-test. *indicates P < 0.05, **P < 0.01 and ***P < 0.001. Each data are from three independent experiments. (C) The protein and mRNA levels of p21 are up-regulated in HSCARG−/− cells. The protein and mRNA levels of p21 were examined in wild-type HCT116 cells, HSCARG−/− HCT116 cells and HSCARG−/− HCT116 cells transfected with Flag-HSCARG. The levels of p53 and p-p53 were also detected in these cells. (D) Deletion of HSCARG leads to a decreased cell growth rate, which can be partially rescued by USP7. Living cells were counted by trypan blue staining at different time points after initial seeding with 5 × 103 cells. (E) FACS analysis showed that HSCARG−/− cells were arrested in the G1 phase. The percentages of cells in different stage of cell cycle were detected by FACS analysis. Values were reported as mean ± SD; P value was determined by Student's t-test. **indicates P < 0.01.
Next, we performed qRT-PCR to compare the mRNA levels of a number of DNA damage repair proteins including DDB2, GADD5, SESN1 and p53R2. The results confirmed that the mRNA levels of ddb2, sesn1 and p53R2 were clearly higher in HSCARG−/− cells than in wild-type HCT116 cells (Figure 6B). Besides, the mRNA level of p21, a critical gene controls G1/S phase transition in response to DNA damage, was three-fold higher in HSCARG−/− cells than that of wild-type HCT116 cells. Western blot and qRT-PCR analysis further confirmed this result (Figure 6C). As p21 activation is often resulted from p53 phosphorylation, we also detected the phosphorylation of p53. The results showed that in HSCARG−/− cells, Ser15 phosphorylation of p53 was indeed higher than that of wild-type HCT116 cells and HSCARG−/− cells reintroduced with HSCARG (Figure 6C). We also noticed that compared to the wild-type cells, HSCARG−/- cells exhibited a clearly reduced growth rate. Because HSCARG depends on USP7 in regulating H2A ubiquitination, we also assessed the effect of USP7 on HSCARG in modulating cell growth. We found that USP7−/− cells showed a slower growth rate. And USP7 partially rescued the growth rate of HSCARG−/− cells, while HSCARG did not rescue the growth rate of USP7−/− cells (Figure 6D). These data suggest that HSCARG regulates cell proliferation partially that depends on USP7.
Furthermore, we investigated the effect of HSCARG on cell cycle. The results showed that deletion of HSCARG resulted in an obvious cell cycle arrest, with cells delayed mainly in the G1 phase (Figure 6E), and this was consistent with higher expression level of p21 in HSCARG−/− cells. Collectively, these results indicate that deletion of HSCARG results in persistent activation of checkpoint signaling and affects G1/S phase transition.
DISCUSSION
In this study, we identified a non-catalytic protein HSCARG as a negative regulator of H2A ubiquitination and found that it affects cell cycle in response to DNA damage. Mechanistically, HSCARG interacts with and depends on the deubiquitinase USP7 to inhibit PRC1 ubiquitination and thereby reduces H2A ubiquitination. Furthermore, we demonstrated that HSCARG functions in DNA damage response by affecting ubiquitination of H2A and localization of repair protein. Knockout of HSCARG causes a persistent activation of checkpoint signaling and cell cycle arrest, which is at least partially due to the lower efficiency of HSCARG in reducing H2A ubiquitination.
RING1, RING2 and BMI1 are all ring domain-containing proteins and previous reports show that the three proteins could be ubiquitinated (20,35,46). Here we found that HSCARG strongly inhibits RING1 ubiquitination, which is independent of its NADPH binding activity. Besides, HSCARG could also inhibit RING2 and BMI1 ubiquitination. Recent studies have shown that USP7 can remove ubiquitin chains from PRC1 (35,46). Our study showed that HSCARG, USP7 and RING1 could form a complex, and HSCARG depends on USP7 in inhibiting PRC1 ubiquitination (Figure 2). USP11 is another deubiquitinase of RING1, which has redundant function with USP7 (35). We also examined whether there is a correlation between HSCARG and USP11. As shown in Supplementary Figure S5, HSCARG does not affect the activity of USP11 and vice versa. These results indicate that HSCARG specifically cooperates with USP7 in inhibiting PRC1 ubiquitination.
Previous studies indicate that ubiquitination of PRC1 plays a critical role in regulating PRC1 catalytic activity (20), we therefore assessed whether HSCARG affects H2A ubiquitination. As expected, HSCARG strongly inhibits H2A, but not H2B ubiquitination (Supplementary Figure S6), which is not dependent on NADPH binding ability as well (Supplementary Figure S1E). Considering USP7 is incapable of deubiquitinating ubiquitinated H2A in vitro (51), and HSCARG does not alter chromatin association of PRC1 (Supplementary Figure S7), we therefore believe that HSCARG affects H2A ubiquitination through regulating the E3 ligase activity of PRC1 by recruiting deubiquitinase USP7. And the observation that USP7 inhibits H2A ubiquitination in cells is likely due to its deubiquitinated activity on PRC1.
Altogether, our study indicates that HSCARG interacts with and depends on USP7 to inhibit PRC1 ubiquitination and reduces its catalytic activity, which subsequently decreases the level of H2A ubiquitination. We present a previously un-described regulatory mechanism of H2A ubiquitination that proteins affecting PRC1 ubiquitination also affect the ubiquitination level of H2A.
Both monoubiquitination and K63-linked polyubiquitination of H2A are important in DNA damage response (2–5,19,20). Adding the first ubiquitin moiety to the substrate is thought to be the limiting step in polyubiquitin chain formation (52). Recent studies have shown that monoubiquitination catalyzed by PRC1 or RNF8 can be extended by RNF168, after which the polyubiquitin chains of H2A act as docking sites for the accumulation of repair proteins in response to DNA damage (15,53). The regulatory function of HSCARG in PRC1 and H2A ubiquitination suggests that HSCARG may also participate in the DNA damage response. Notably, we found that HSCARG is involved in these processes. HSCARG inhibits IR-induced H2A ubiquitination and abolishes the accumulation of RAP80 into IR-induced foci without affecting H2AX phosphorylation (Figure 5).
In response to DNA damage, parallel to the recruitment of repair proteins, checkpoint signaling is activated to induce cell cycle arrest (54). After the lesion points are repaired, the ubiquitin chains of H2A ought to be removed and then cells will re-enter the normal cell cycle. Here we found that in HSCARG−/− cells, removal of H2A ubiquitin chains decreases, which leads to trap of repair protein at lesion points and persistent activation of checkpoint signaling such as phosphorylation of CHK2 and elevated expressions of the DNA damage response proteins, and finally results in the slower growth rate of HSCARG−/− cells.
Collectively, our study has uncovered a novel function of HSCARG as an inhibitor of the E3 ubiquitin ligase activity of PRC1 and a negative regulator of H2A ubiquitination, and has established a role for HSCARG in DNA damage response. This study may explain, at least partially, how regulator of H2A ubiquitination is linked to the cell cycle arrest and affects cell proliferation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGMENTS
We thank Dr Hengbin Wang at University of Alabama at Birmingham for the gift of plasmids of RING1, RING2, BMI1, Dr Goedele Maertens at Cancer Research UK for USP7, Dr Xiaochun Yu at University of Michigan Medical School for RAP80, Dr Zhijian Chen at University of Texas Southwestern Medical Center for ubiquitin, Dr Xiaojie Tan and Jianhua Yang at Baylor College of Medicine for USP11 and USP11 shRNA and Dr Bert Vogelstein at Ludwig Center, Johns Hopkins, for providing USP7−/− HCT116 cell line.
FUNDING
National Science Foundation of China [30930020 and 31170709]; National High Technology and Development Program of China 973 programs [2010CB911800]; Doctoral Fund of Ministry of Education of China [20130001130003]; International Centre for Genetic Engineering and Biotechnology [CRP/CHN09-01]. Funding for open access charge: National Science Foundation of China [30930020 and 31170709]; National High Technology and Development Program of China 973 programs [2010CB911800]; Doctoral Fund of Ministry of Education of China [20130001130003]; International Centre for Genetic Engineering and Biotechnology [CRP/CHN09-01].
Conflict of interest statement. None declared.
REFERENCES
- 1.Goldknopf I.L., Taylor C.W., Baum R.M., Yeoman L.C., Olson M.O., Prestayko A.W., Busch H. Isolation and characterization of protein A24, a ‘histone-like’ non-histone chromosomal protein. J. Biol. Chem. 1975;250:7182–7187. [PubMed] [Google Scholar]
- 2.Wang H., Wang L., Erdjument-Bromage H., Vidal M., Tempst P., Jones R.S., Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 3.Cao R., Tsukada Y., Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Mailand N., Bekker-Jensen S., Faustrup H., Melander F., Bartek J., Lukas C., Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 5.Huen M.S., Grant R., Manke I., Minn K., Yu X., Yaffe M.B., Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leeb M., Wutz A. Ring1B is crucial for the regulation of developmental control genes and PRC1 proteins but not X inactivation in embryonic cells. J. Cell Biol. 2007;178:219–229. doi: 10.1083/jcb.200612127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richly H., Di Croce L. The flip side of the coin: role of ZRF1 and histone H2A ubiquitination in transcriptional activation. Cell Cycle. 2011;10:745–750. doi: 10.4161/cc.10.5.14795. [DOI] [PubMed] [Google Scholar]
- 8.Richly H., Rocha-Viegas L., Ribeiro J.D., Demajo S., Gundem G., Lopez-Bigas N., Nakagawa T., Rospert S., Ito T., Di Croce L. Transcriptional activation of polycomb-repressed genes by ZRF1. Nature. 2010;468:1124–1128. doi: 10.1038/nature09574. [DOI] [PubMed] [Google Scholar]
- 9.Vissers J.H., Nicassio F., van Lohuizen M., Di Fiore P.P., Citterio E. The many faces of ubiquitinated histone H2A: insights from the DUBs. Cell Div. 2008;3:8. doi: 10.1186/1747-1028-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakagawa T., Kajitani T., Togo S., Masuko N., Ohdan H., Hishikawa Y., Koji T., Matsuyama T., Ikura T., Muramatsu M., et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008;22:37–49. doi: 10.1101/gad.1609708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou W., Zhu P., Wang J., Pascual G., Ohgi K.A., Lozach J., Glass C.K., Rosenfeld M.G. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol. Cell. 2008;29:69–80. doi: 10.1016/j.molcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X.Y., Varthi M., Sykes S.M., Phillips C., Warzecha C., Zhu W., Wyce A., Thorne A.W., Berger S.L., McMahon S.B. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol. Cell. 2008;29:102–111. doi: 10.1016/j.molcel.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shao G., Lilli D.R., Patterson-Fortin J., Coleman K.A., Morrissey D.E., Greenberg R.A. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc. Natl. Acad. Sci. U.S.A. 2009;106:3166–3171. doi: 10.1073/pnas.0807485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang B., Elledge S.J. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2007;104:20759–20763. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doil C., Mailand N., Bekker-Jensen S., Menard P., Larsen D.H., Pepperkok R., Ellenberg J., Panier S., Durocher D., Bartek J., et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 16.Joo H.Y., Zhai L., Yang C., Nie S., Erdjument-Bromage H., Tempst P., Chang C., Wang H. Regulation of cell cycle progression and gene expression by H2A deubiquitination. Nature. 2007;449:1068–1072. doi: 10.1038/nature06256. [DOI] [PubMed] [Google Scholar]
- 17.Nicassio F., Corrado N., Vissers J.H., Areces L.B., Bergink S., Marteijn J.A., Geverts B., Houtsmuller A.B., Vermeulen W., Di Fiore P.P., et al. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr. Biol. 2007;17:1972–1977. doi: 10.1016/j.cub.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 18.Joo H.Y., Jones A., Yang C., Zhai L., Smith A.D.t., Zhang Z., Chandrasekharan M.B., Sun Z.W., Renfrow M., Wang Y., et al. Regulation of histone H2A and H2B deubiquitination and Xenopus development by USP12 and USP46. 2010;286:7190–7201. doi: 10.1074/jbc.M110.158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei J., Zhai L., Xu J., Wang H. Role of Bmi1 in H2A ubiquitylation and Hox gene silencing. J. Biol. Chem. 2006;281:22537–22544. doi: 10.1074/jbc.M600826200. [DOI] [PubMed] [Google Scholar]
- 20.Ben-Saadon R., Zaaroor D., Ziv T., Ciechanover A. The polycomb protein Ring1B generates self atypical mixed ubiquitin chains required for its in vitro histone H2A ligase activity. Mol. Cell. 2006;24:701–711. doi: 10.1016/j.molcel.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 21.Zaaroor-Regev D., de Bie P., Scheffner M., Noy T., Shemer R., Heled M., Stein I., Pikarsky E., Ciechanover A. Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc. Natl. Acad. Sci. U.S.A. 2010;107:6788–6793. doi: 10.1073/pnas.1003108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berkovich E., Monnat R.J., Jr, Kastan M.B. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 2007;9:683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 24.Falck J., Coates J., Jackson S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 25.Kim H., Chen J., Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- 26.Wu J., Huen M.S., Lu L.Y., Ye L., Dou Y., Ljungman M., Chen J., Yu X. Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol. Cell. Biol. 2009;29:849–860. doi: 10.1128/MCB.01302-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R., III, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y., et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 28.Jazayeri A., Falck J., Lukas C., Bartek J., Smith G.C., Lukas J., Jackson S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 29.Smith J., Tho L.M., Xu N., Gillespie D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 30.Ahn J.Y., Schwarz J.K., Piwnica-Worms H., Canman C.E. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–5936. [PubMed] [Google Scholar]
- 31.Stracker T.H., Usui T., Petrini J.H. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst) 2009;8:1047–1054. doi: 10.1016/j.dnarep.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng X., Dai X., Zhao Y., Chen Q., Lu F., Yao D., Yu Q., Liu X., Zhang C., Gu X., et al. Restructuring of the dinucleotide-binding fold in an NADP(H) sensor protein. Proc. Natl. Acad. Sci. U.S.A. 2007;104:8809–8814. doi: 10.1073/pnas.0700480104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gan Q., Li T., Hu B., Lian M., Zheng X. HSCARG inhibits activation of NF-kappaB by interacting with IkappaB kinase-beta. J. Cell Sci. 2009;122:4081–4088. doi: 10.1242/jcs.054007. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J., Bai D., Ma X., Guan J., Zheng X. hCINAP is a novel regulator of ribosomal protein-HDM2-p53 pathway by controlling NEDDylation of ribosomal protein S14. 2012;33:246–254. doi: 10.1038/onc.2012.560. [DOI] [PubMed] [Google Scholar]
- 35.Maertens G.N., El Messaoudi-Aubert S., Elderkin S., Hiom K., Peters G. Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. EMBO J. 2010;29:2553–2565. doi: 10.1038/emboj.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wysocka J., Reilly P.T., Herr W. Loss of HCF-1-chromatin association precedes temperature-induced growth arrest of tsBN67 cells. Mol. Cell. Biol. 2001;21:3820–3829. doi: 10.1128/MCB.21.11.3820-3829.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang J., Huen M.S., Kim H., Leung C.C., Glover J.N., Yu X., Chen J. RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat. Cell Biol. 2009;11:592–603. doi: 10.1038/ncb1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao Y., Zhang J., Li H., Li Y., Ren J., Luo M., Zheng X. An NADPH sensor protein (HSCARG) down-regulates nitric oxide synthesis by association with argininosuccinate synthetase and is essential for epithelial cell viability. J. Biol. Chem. 2008;283:11004–11013. doi: 10.1074/jbc.M708697200. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X., Guo C., Chen Y., Shulha H.P., Schnetz M.P., LaFramboise T., Bartels C.F., Markowitz S., Weng Z., Scacheri P.C., et al. Epitope tagging of endogenous proteins for genome-wide ChIP-chip studies. Nat. Methods. 2008;5:163–165. doi: 10.1038/nmeth1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satijn D.P., Gunster M.J., van der Vlag J., Hamer K.M., Schul W., Alkema M.J., Saurin A.J., Freemont P.S., van Driel R., Otte A.P. RING1 is associated with the polycomb group protein complex and acts as a transcriptional repressor. Mol. Cell. Biol. 1997;17:4105–4113. doi: 10.1128/mcb.17.7.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Satijn D.P., Otte A.P. RING1 interacts with multiple Polycomb-group proteins and displays tumorigenic activity. Mol. Cell. Biol. 1999;19:57–68. doi: 10.1128/mcb.19.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu M., Li P., Li M., Li W., Yao T., Wu J.W., Gu W., Cohen R.E., Shi Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- 43.Komander D., Clague M.J., Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 44.Reyes-Turcu F.E., Ventii K.H., Wilkinson K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai X., Li Y., Meng G., Yao S., Zhao Y., Yu Q., Zhang J., Luo M., Zheng X. NADPH is an allosteric regulator of HSCARG. J. Mol. Biol. 2009;387:1277–1285. doi: 10.1016/j.jmb.2009.02.049. [DOI] [PubMed] [Google Scholar]
- 46.de Bie P., Zaaroor-Regev D., Ciechanover A. Regulation of the Polycomb protein RING1B ubiquitination by USP7. Biochem. Biophys. Res. Commun. 2010;400:389–395. doi: 10.1016/j.bbrc.2010.08.082. [DOI] [PubMed] [Google Scholar]
- 47.Zhou W., Wang X., Rosenfeld M.G. Histone H2A ubiquitination in transcriptional regulation and DNA damage repair. Int. J. Biochem. Cell Biol. 2009;41:12–15. doi: 10.1016/j.biocel.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 48.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 49.Osley M.A., Fleming A.B., Kao C.F. Histone ubiquitylation and the regulation of transcription. Results Probl. Cell Differ. 2006;41:47–75. doi: 10.1007/400_006. [DOI] [PubMed] [Google Scholar]
- 50.Wright D.E., Wang C.Y., Kao C.F. Histone ubiquitylation and chromatin dynamics. Front. Biosci. 2011;17:1051–1078. doi: 10.2741/3973. [DOI] [PubMed] [Google Scholar]
- 51.van der Knaap J.A., Kumar B.R., Moshkin Y.M., Langenberg K., Krijgsveld J., Heck A.J., Karch F., Verrijzer C.P. GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Mol. Cell. 2005;17:695–707. doi: 10.1016/j.molcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 52.Deshaies R.J., Joazeiro C.A. RING domain E3 ubiquitin ligases. Ann. Rev. Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 53.Gieni R.S., Ismail I.H., Campbell S., Hendzel M.J. Polycomb group proteins in the DNA damage response: a link between radiation resistance and ‘stemness’. Cell Cycle. 2011;10:883–894. doi: 10.4161/cc.10.6.14907. [DOI] [PubMed] [Google Scholar]
- 54.Bartek J., Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.