

Abstract

To develop effective oral treatment for multiple sclerosis (MS), we discovered a series of alkyl-substituted biaryl amino alcohols as selective S1P1 modulators. One exemplar is (S)-2-amino-2-(5-(4-(octyloxy)-3-(trifluoromethyl)phenyl)-1,3,4-thiadiazol-2-yl)propan-1-ol (10, GSK1842799). Upon phosphorylation, the compound (10-P) showed subnanomole S1P1 agonist activity with >1000× selectivity over S1P3. The alcohol 10 demonstrated good oral bioavailability and rapid in vivo conversion to 10-P. Dosed orally at 0.1 mg/kg, 10 significantly reduced blood lymphocyte counts 6 h postdose, and at 3 mg/kg, 10 achieved efficacy equivalent to FTY720 in the mouse EAE model of MS. Further pharmacokinetic/pharmacodynamic (PK/PD) study with cynomolgus monkeys indicated that, after oral dosing of 10 at 3.8 mg/kg, the active phosphate reached plasma levels that are comparable to FTY-720 phosphate (FTY-P) revealed in human clinical pharmacokinetics studies. On the basis of the favorable in vitro ADME and in vivo PK/PD properties as well as broad toxicology evaluations, compound 10 (GSK1842799) was selected as a candidate for further clinical development.
Keywords: S1P1 modulator, biaryl aminoalcohol, prodrug, multiple sclerosis, mouse EAE model
Chronic demyelination and scarring of the neuronal cells in brain, spinal cord, and central nervous system lead to a debilitating neurodegenerative autoimmune disorder known as multiple sclerosis (MS). This disease has an onset usually in young adults and more commonly in females, affecting lives of hundreds and thousands of people across the globe.1 This inflammatory disorder poses diverse neurological symptoms often leading to physical and cognitive disability.2 The emergence of FTY-720 (Fingolimod) as a novel class of orally active immune-modulating agents for multiple sclerosis and solid organ transplant rejection prevention has established the sphingosine-1-phosphate (S1P) receptor class of G-protein coupled receptors (GPCRs) as a competitive and promising field of research and development. FTY-720 is a prodrug that, upon in vivo phosphorylation, activates the S1P1 receptor and sequesters lymphocytes in lymph nodes, preventing them from trafficking to lymphoid tissues and contributing to autoimmune reactions.3 FTY-720 reduces both relapses and disability progression in relapsing-remitting multiple sclerosis (RRMS) patients.4−7 However, aside from the clinical benefits of FTY-720, a number of adverse side effects such as bradycardia, skin cancer, liver injury, infections, and increased blood pressure were attributed to activity on S1P3, leaving a window of opportunity for exploration of alternative chemotypes with improved overall profiles.
Recently, we published a number of novel S1P1 agonists, a few of which fall within the same class of prodrug as FTY-720 that require in vivo phosphorylation in order to trigger any effects on S1P receptors (Table 1).8−11 The phosphorylation of prodrug FTY-720 to its active drug FTY-720-monophosphate is facilitated by sphingosine kinase 2 (SphK2).12 One major challenge in the prodrug strategy in S1P activation is its in vivo phosphorylation of the prodrug to the desired phospho-drug since this process requires prodrug phosphorylation with either SphK1 or SphK2, while the phospho-drug could be converted back to the starting prodrug through the action of sphingosine-1-phosphate phosphatase 1 (S1PP1), sphingosine-1-phosphate phosphatase 2 (S1PP2), or a potential lipid phosphate phosphatase.13 In our most recent prodrug publication,10 we reported on preclinical candidate PPI-4955 (Table 1) and its phosphorylation outcome in rodent models. In an effort to establish a backup molecule to PPI-4955 with improved profile especially in in vivo phosphorylation, we carried out further structure–activity relationship (SAR) studies with a focus on sphingosine kinase activity and in vivo phosphorylation relationships. Since the PPI-4955 series are structurally homologous to FTY-720 and human SphK2 is known to phosphorylate FTY-720 30-fold better than SphK1,14 our structural modifications were mainly driven by SphK2 activity. Herein, we report our compound design and synthesis as well as in vitro and in vivo activity data that led to the discovery of clinical development candidate GSK1842799.
Table 1. Effects of Head-Piece Modification on SphK2 Activity, in Vivo Phosphorylation, and S1P1 Activity and Selectivity.
| phosphate | R | S1P1 (nM)a | S1P3 (nM)a | selectivity (S1P1/ S1P3) | prodrug | mSphK2 (%FTY) | hSphK2 (%FTY) | % in vivo PO4 (mice)b |
|---|---|---|---|---|---|---|---|---|
| FTY-720-P | 1.20 | 13.3 | 11 | FTY-720 | 100 | 100 | 70 | |
| PPI-4955-P | CH3 | 0.83 | 7352 | 8862 | PPI-4955 | 49.6 | 7.8 | 59 |
| 1-P | H | 0.30 | 650 | 2166 | 1 | 5.2 | 2.8 | 3.0 |
| 2-P | CH2OH | 2.3 | >10000 | >4348 | 2 | 1.9 | 1.9 | 20 |
[33P] binding activity on S1P1 and S1P3–5 receptor subtypes.
Percent conversion of alcohol (prodrug) to phosphate (drug) at 6 h dose administration upon 10 mg/kg oral (PO) administration of the alcohol.
Our SAR study started with the modification of the head-piece amino-alcohol and the impact on both human SphK2 (hSphK2) and mouse SphK2 (mSphK2) activity with respect to PPI-4955. The biological data for PPI-4955 and head-piece modified analogues 1 and 2 as well as FTY-720 (as control) are reported in Table 1. In pursuit of better understanding PPI-4955 and analogue activity as substrates of SphK2 and the correlation of this data with in vivo phosphorylation, all the compounds were tested for in vitro activity against mSphK2 and hSphK2 as well as in vivo conversion to the corresponding phosphates in mouse upon oral administration. In comparison to FTY-720 phosphate (FTY-720-P), PPI-4955 phosphate (PPI-4955-P) showed better potency and much improved selectivity for S1P1 over S1P3. Setting FTY-720 activity against mSphK2 and hSphK2 at 100%, PPI-4955 performed reasonably well in mSphK2 activity (about 50%) correlating to mouse in vivo phosphorylation of 59% compared to FTY-720 of 70%. However, activity of PPI-4955 against hSphK2 was low (7.8%) with respect to activity of FTY-720, indicating a difference between mouse and human SphK2 at the substrate binding site and a potential point for further improvement in SphK2 activity. The first attempt at modification of the head-piece, where the R group was changed either into a proton (1) or a hydroxymethyl (2), led to decreased SphK2 activities. However, phosphates of both compounds still showed potent S1P1 binding activity and good selectivity against S1P3.
Chemical modification of the PPI-4955 imidazole ring was then explored. Similarly, all of the corresponding compounds were tested for in vitro activity as potential substrates for mSphK2 and hSphK2 as well as in vivo phosphorylation in mouse upon oral administration. A quick profiling of the azole compounds generated provided insight and guidance in further SAR exploration (Table 2). As S1P receptor subtype binding data indicated, all the corresponding phosphates for compounds 3–5 and 6–8 maintained excellent S1P1 binding activity and analogous selectivity for S1P1 over S1P3 as PPI-4995-phosphate. For sphingosine kinase activity, while the added 4-methyl group on the imidazole ring (3) was detrimental for both mouse and human SphK2 enzymes, the corresponding 5-methyl oxazole analogue (4) gave a similar profile as that for PPI-4955. The 2,4-disubstituted thiazole (5) and 2,5-disubsituted oxazole (6) analogues were less active than imidazole-containing PPI-4955. Interestingly, the 2,5-substituted thiazole (7) and 1,3,4-thiadiazole (8) showed much improved activity against both mSphK2 and hSphK2 enzymes, suggesting the SphK2 enzyme activity is very sensitive to the azole moiety modifications where a 2,5-disubstituted thiazole (7) and thiadiazole (8) orientations are analogous to a para-subtituted phenyl moiety, while the 2,4-disubstituted thiazole (5) and 2,5-disubsituted oxazole (6) analogues are closer to a meta-substituted phenyl group. Accordingly, the corresponding in vivo phosphorylation levels were also significantly increased (89% and 90%, respectively), a demonstration of the correlation between SphK2 enzyme activity and the in vivo phosphorylation.
Table 2. Structural Modification of PPI-4955 Imidazole Ring Effects on Agonist Profile.
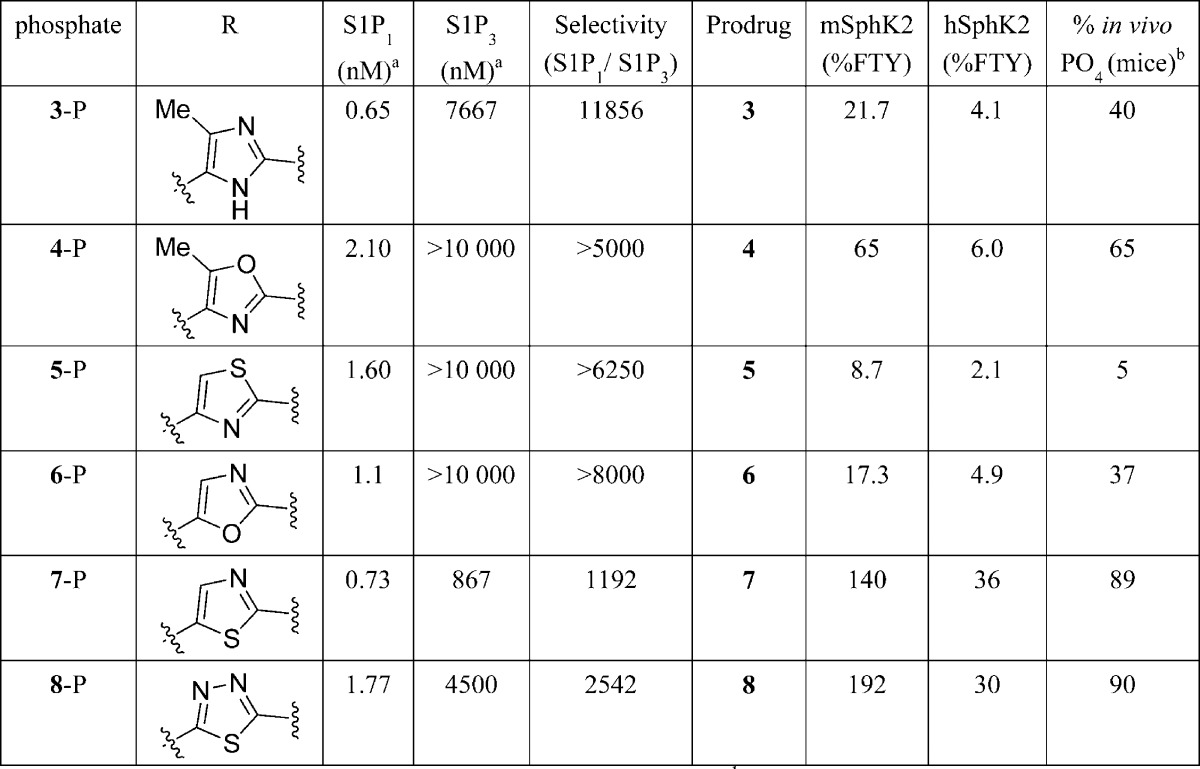
[33P] binding activity on S1P1 and S1P3–5 receptor subtypes.
Percent conversion of alcohol (prodrug) to phosphate (drug) at 6 h dose administration upon 10 mg/kg oral (PO) administration of the alcohol.
The findings in 2,5-disubstituted thiazole 7 and disubstituted-1,3,4-thiadiazole 8 provided the basis and rationale for the next generation agonist design. Previously, it was observed that both FTY-720 and a number of prodrugs with the alkyl tail group, analogous to FTY-720, have demonstrated good mSphK2 and in vivo lymphopenia activity. It was hypothesized that replacement of the tail group in both 7 and 8 with an alkyl group might lead to further improvement in SphK2 activity. As per earlier reports,8 the best alkyl tail group with improved lead prodrug profile was octane ether; thus, a series of azole analogues with the n-octyl tail group were synthesized as shown in Table 3. Investigation of SphK2 activity of the prodrugs with tail group and azole ring modifications proved to be instrumental in directing the research effort toward the next generation of the lead molecules. As demonstrated in Table 3, replacement of the [1,1′-biphenyl]-4-ylmethyl ether group with an octyl ether in both 7-P and 8-P gave compounds 9-P and 10-P, which showed a good improvement in S1P1 binding activity, while still maintaining excellent selectivity for S1P1 over S1P3. At the same time, both prodrugs 9 and 10 showed a substantial improvement in both mSphK2 (2-fold) and hSphK2 (5-fold) over the predecessors 7 and 8. Both prodrugs 9 and 10 gave analogous lymphopenia profile as FTY720 with quantitative in vivo phosphoryaltion when administered orally in mouse. Further modification of the azole system to oxadiazole 11-P, pyrazole 12-P, isoxazole 13-P, oxadiazole 14-P, and imidazole 15-P provided comparable S1P1 binding profile as the compounds 9-P and 10-P, while no further improvement was observed in SphK2 activity, especially hSphK2 activity. It was interesting to see that, while the 1,3-thiazole (9) and 1,3,4-thiadiazole (10) analogues gave the best SphK2 activity profile, the 1,3,4-oxodiazole (11) and imidazole (15) analogues were poor substrates for SphK2 in the series, suggesting that the insertion of a sulfur atom in the azole ring might have changed the geometry of the molecule significantly.
Table 3. mSphK2 and hSphK2 Activity of Prodrug Analogues Containing Octyl Ether Tail.
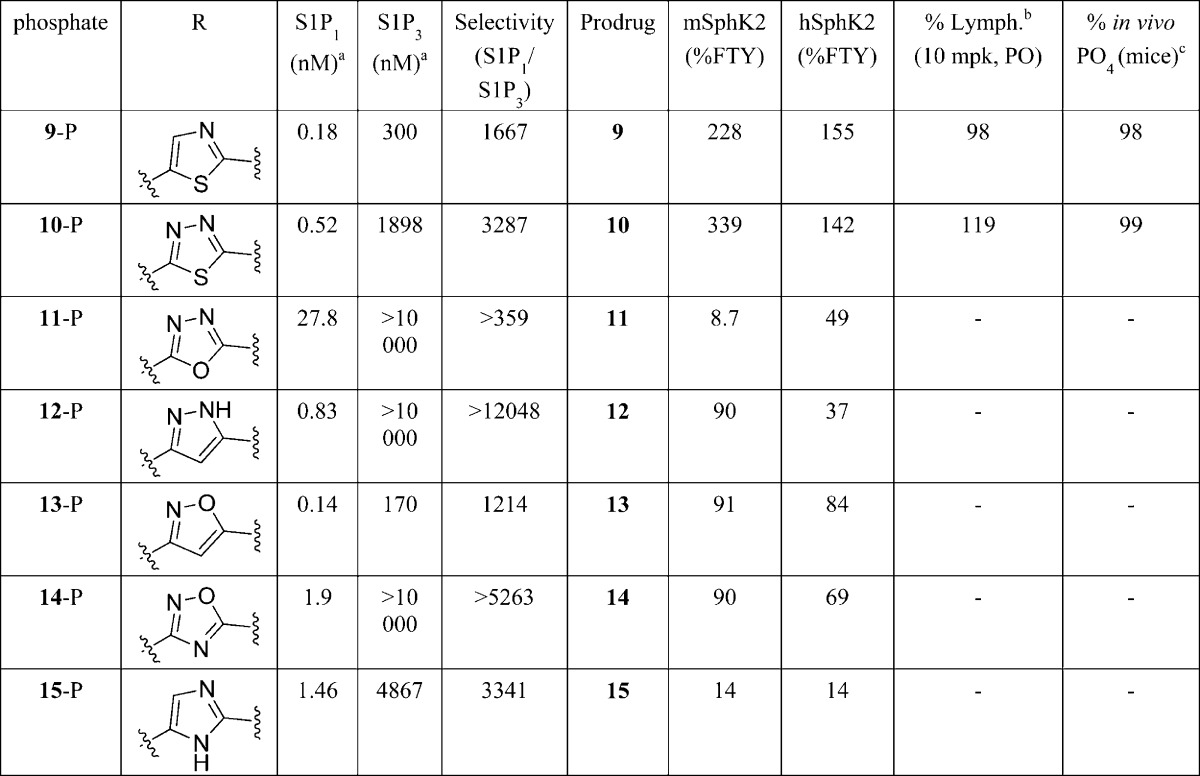
[33P] binding activity on S1P1 and S1P3–5 receptor subtypes.
Percent lymphopenia observed in mouse 6 h postdose upon 10 mg/kg oral (PO) administration of the corresponding alcohol in comparison to oral administration of 1 mg/kg of FTY-720.
Percent conversion of alcohol (prodrug) to phosphate (drug) at 6 h dose administration upon 10 mg/kg oral (PO) administration of the alcohol.
In order to confirm agonist activity and selectivity of the corresponding phosphates, compounds 9-P and 10-P were further investigated in a [γ-35S]GTP functional assay15 as reported in Table 4. The two compounds showed analogous activity profiles toward all of the S1P receptor subtypes with subnanomolar agonist affinity on S1P1. Neither of the agonists had any binding effect on S1P3, demonstrating an excellent selectivity for S1P1 over S1P3. Notably, both compounds also had potent agonist activity on S1P4 and S1P5.
Table 4. [γ-35S]GTP Functional Activity of 9-P and 10-P on all S1P Receptor Subtypes.
| agonist | X | hS1P1 EC50 (nM) | hS1P2 EC50 (nM) | hS1P3 EC50 (nM) | hS1P4 EC50 (nM) | hS1P5 EC50 (nM) | S1P3/S1P1 |
|---|---|---|---|---|---|---|---|
| S1P | 4.25 | 1.3 | 3.94 | 13 | 1.8 | 0.9 | |
| 9-P | CH | 0.44 | >3000 | 3.60 | 2.00 | >6800 | |
| 10-P | N | 0.26 | >3000 | 4.57 | 0.66 | >11500 |
The lead prodrugs 9 and 10 were evaluated in an in vivo dose–response analysis when orally administered in mice (Figure 1). At the lowest dose of 0.1 mg/kg, both prodrugs showed a mild reduction in lymphocyte counts, while a significant lymphopenia was observed in all three higher doses. Both compounds showed excellent dose responsiveness when administered orally at doses of 0.1, 0.3, 1.0, and 3.0 mg/kg. The prodrugs 9 and 10 were further evaluated in the MOG peptide experimental autoimmune encephalitis (EAE) mouse multiple sclerosis disease model (Figure 2).16 In this experiment, both compounds demonstrated efficacy equivalent to FTY-720 when administered orally once a day at 3 mg/kg, nearly completely reversing the signs and symptoms of disease (Figure 2).
Figure 1.
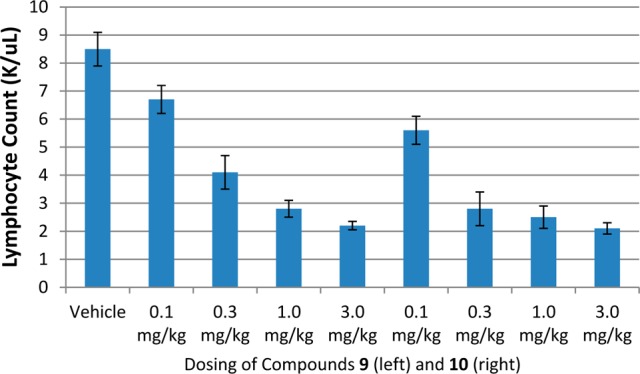
Dose response lymphopenia of lead compounds 9 and 10 relative to the vehicle.
Figure 2.
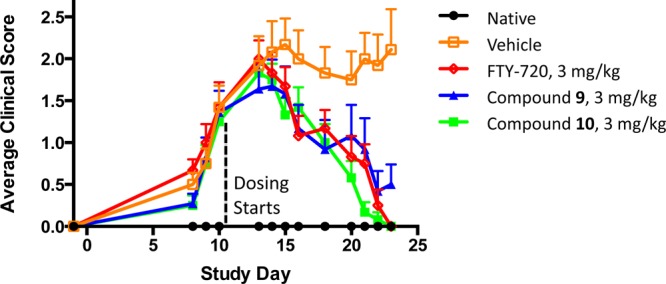
Effect of lead S1P1 agonists 9 and 10 in the MOG peptide EAE model of multiple sclerosis.
In vitro ADME profiles of 9 and 10 met general criteria for a development candidate with reasonable aqueous solubility and protein binding, high permeability in Caco-2 with no efflux, no CYP450 inhibition, and good metabolic stability (Table S1, Supporting Information). An in vivo dog PK study suggested phosphate drug exposure, half-life, and oral bioavailability (Table S2, Supporting Information) that support once a day dosing. When dosed orally (single-dose) in multiple species (Table S3, Supporting Information), the active phosphate of 10-P reached plasma levels that ranged ∼10–50 ng/mL. In cynomolgus monkeys administered a dose of 3.8 mg/kg, a plasma Cmax = 50.9 ng/mL was achieved and a lymphocytosis EC90 = 16.2 ng/mL, which is comparable to FTY-720 phosphate levels revealed in human clinical pharmacokinetics studies.17,18 After broad toxicology evaluations, compound 10 (GSK1842799) was selected as a candidate for further development.
Syntheses of the above compounds are outlined in Schemes 1–3. In Scheme 1, analogously to the synthesis of PPI-4955,10 the substituted bromo-acetophenones (17) were converted to corresponding esters (21) by reacting with different head-piece moieties including Boc-serine–OH, 5-(tert-butoxycarbonylamino)-2,2-dimethyl-1,3-dioxane-5-carboxylic acid (18), 5-((tert-butoxycarbonyl)amino)-2,2-dimethyl-1,3-dioxane-5-carboxylic acid (19), and (S)-3-(tert-butoxycarbonyl)-2,2,4-trimethyloxazolidine-4-carboxylic acid (20). Using NH4OAc under toluene refluxing conditions, these esters were cyclized to substituted imidazole or oxazole intermediates, which upon acidic treatment provided final compounds 1, 2, 3, 4, and 15. Meanwhile, the head-piece (20) was first converted to amide then to thioamide (23) by Lawesson’s reagent under THF refluxing condition. The thioamide further reacted with bromo-acetophenone (17, R1 = H) to form 2,4-disubstituted thiazole (23), which afforded the desired compound 5 after removal of the protecting group.
Scheme 1. Synthesis of Imidazole, Oxazole, and Thiazole Analogues (1–5 and 15).

Reagents and conditions: (a) CuBr2, EtOAc/CHCl3 (1:1), reflux; (b) Cs2CO3, DMF, 18, 19, or 20; (c) NH4OAc, toluene, reflux; (d) 6 N HCl, THF or TFA, DCM; or TsOH, MeOH, reflux; (e) 22, THF, reflux.
Scheme 3. Synthesis of Pyrazole, Isoxazole, and 1,2,4-Oxadiazole Analogues 12–14.

Reagents and conditions: (a) N,O-dimethylhydroxylamine hydrochloride, HATU, DIPEA, DMF; (b) MeLi, THF, −78 °C; (c) 26 (R = n-heptyl), (COCl)2, DCM; (d) LiHMDS, THF, −78–0 °C; (e) hydrazine, MeOH; (f) hydroxylamine hydrochloride, pyridine; (g) TFA, DCM; (h) n-octyl–OH, KOtBu, THF, 70 °C; (i) hydroxylamine hydrochloride, EtOH, Na2CO3, H2O; (j) 20, HATU, DIPEA, DCM/DMF; (k) DMF, 120 °C; (l) TFA, DCM.
In Scheme 2, the intermediate 25, prepared from bromo-acetophenone (17) through azide and amine intermediates followed by coupling with the orthogonally protected α-methyl serine (20) or from 3-trifluoromethyl-4-fluorobenzoic acid (26) via hydramide formation and coupling reaction with 20, was cyclized using Lawesson’s reagent either in toluene or dichloromethane to afford thiazole and thiadiazole analogues 7, 8, 9, and 10 after deprotection with TFA. This synthetic route was developed into a fully telescoped process synthesis for compound 10 at multikilogram scale.19 Alternatively, the intermediates 25 were converted to oxazole 6 or 1,3,4-oxadiazole 11 using triphenylphosphine and hexachloroethane or triphenylphosphine and trichloroacetonitrile correspondingly after deprotection with TFA.
Scheme 2. Synthesis of Oxazole, Thiazole, Thiadiazole, and 1,3,4-Oxadiazole Analogues 6–11.

Reagents and conditions: (a) (i) CuBr2, EtOAc/CHCl3 (1:1), reflux; (ii) NaN3, DMF; (b) H2 (gas), 5% Pd–C, MeOH, HCl; (c) 20, HATU, DIPEA, DCM/DMF; (d) R2CH2OH, KOtBu, THF, 70 °C; (e) hydrazine, HATU, DIPEA, DCM/DMF; (f) Lawesson’s reagent, toluene, 120 °C; (g) Lawesson’s reagent, DCM; (h) PPh3 (5 equiv), hexachloroethane (C2Cl6, 2.5 equiv), TEA (10 equiv); (i) PPh3, CCl3CN, CH3CN, MW, 120 °C, 20 min; (j) 6 N HCl, dioxane or TFA, DCM; or TsOH, MeOH, reflux.
In Scheme 3, the head piece building block 20 was converted to methylketone 29 through Weinreb ketone synthesis. The methylketone 29 was then reacted with acid 26 through acid chloride intermediate to give diketone 30, which further condensed with either hydrazine or hydroxylamine followed by deprotection with TFA affording the desired pyrazole and isoxazole analogues 12 and 13. Meanwhile, using 4-fluoro-3-(trifluoromethyl)benzonitrile (32) as starting material, the 1,2,4-oxadiazole analogue 14 was prepared through the formation of hydroxylamidine intermediates 33 and 34 followed by cyclization and Boc deprotection. All of the phosphates were synthesized from corresponding alcohols analogously to the synthesis of PPI-4955-P.10
In summary, replacement of the imidazole ring of the novel S1P1 prodrug PPI-4955 with a series of bioisosteric heterocycles (azoles) and switching the biphenyl moiety with an n-octyl tail led to the identification of several potent and selective prodrugs for modulating S1P1 receptors. Among them, compound 10 demonstrated good in vitro ADME and in vivo PK properties. Upon phosphorylation, it showed subnanomole S1P1 agonist activity with >1000× selectivity over S1P3. Dosed orally at 0.1 mg/kg, 10 significantly reduced blood lymphocyte counts 6 h postdose and achieved in vivo efficacy analogous to FTY720 in the mouse EAE model of MS. Further pharmacokinetic/pharmacodynamic (PK/PD) study with monkeys indicated that oral dosing of 10 at 3.8 mg/kg led to active phosphate species that reached plasma levels comparable to FTY-720 phosphate revealed in human clinical pharmacokinetics studies. Compound 10 (GSK1842799) was selected as a candidate for clinical development.
Supporting Information Available
Experimental details for the synthesis of all the compounds and in vitro ADME and in vivo PK/PD data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ GlaxoSmithKline, Platform Technology & Science, MDR Boston, 830 Winter Street, Waltham, Massachusetts 02451, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Compston A.; Coles A. Multiple Sclerosis. Lancet 2008, 372, 1502–1517. [DOI] [PubMed] [Google Scholar]
- Compston A.; Coles A. Multiple Sclerosis. Lancet 2002, 359, 1221–1231. [DOI] [PubMed] [Google Scholar]
- Brinkmann V.; Billich A.; Baumruker T.; Heining P.; Schmouder R.; Francis G.; Aradhye S.; Burtin P. Fingolimod (FTY720): Discovery and Development of an Oral Drug to Treat Multiple Sclerosis. Nat. Rev. 2010, 9, 883–897. [DOI] [PubMed] [Google Scholar]
- Fujino M.; Funeshima N.; Kitazawa Y.; Kimura H.; Amemiya H.; Suzuki S.; Li X. K. Amelioration of Experimental Autoimmune Encephalomyelitis in Lewis Rats by FTY720 Treatment. J. Pharmacol. Exp. Ther. 2003, 305, 70–77. [DOI] [PubMed] [Google Scholar]
- Kappos L.; Antel J.; Comi G.; Montalban X.; O’Connor P.; Polman C. H.; Haas T.; Korn A. A.; Karlsson G.; Radue E. W. Oral Fingolimod (FTY720) for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2006, 355, 1124–1140. [DOI] [PubMed] [Google Scholar]
- O’Connor P.; Comi G.; Montalban X.; Antel J.; Radue E. W.; de Vera A.; Pohlmann H.; Kappos L. Oral Fingolimod (FTY720) in Multiple Sclerosis: Two-Year Results of a Phase II Extension Study. Neurology 2009, 72, 73–79. [DOI] [PubMed] [Google Scholar]
- Novartis. FTY720 FREEDOMS Study: Initial Results. 2009; see http://www.novartis.com/newsroom/news/2009-09-30_spotlight-fty720.shtml.
- Evindar G.; Bernier S. G.; Kavarana M. J.; Doyle E.; Lorusso J.; Kelley M. S.; Halley K.; Hutchings A.; Wright A. D.; Saha A. K.; Hannig G.; Morgan B. A.; Westlin W. F. Synthesis and Evaluation of Alkoxy-phenylamides and Alkoxy-phenylimidazoles as Potent Sphingosine-1-phosphate Receptor Subtype-1 Agonists. Bioorg. Med. Chem. Lett. 2009, 19, 369–372. [DOI] [PubMed] [Google Scholar]
- Evindar G.; Satz A. L.; Bernier S. G.; Kavarana M. J.; Doyle E.; Lorusso J.; Halley K.; Hutchings A.; Kelley M. S.; Wright A. D.; Saha A. K.; Hannig G.; Morgan B. A.; Westlin W. F. Synthesis and Evaluation of Arylalkoxy- and Biarylalkoxy-phenylamide and Phenylimidazoles as Potent and Selective Sphingosine-1-phosphate receptor Subtype-1 Agonists. Bioorg. Med. Chem. Lett. 2009, 19, 2315–2319. [DOI] [PubMed] [Google Scholar]
- Evindar G.; Bernier S. G.; Doyle E.; Kavarana M. J.; Satz A. L.; Lorusso J.; Blanchette H. S.; Saha A. K.; Hannig G.; Morgan B. A.; Westlin W. F. Exploration of Amino Alcohol Derivatives as Novel, Potent, and Highly Selective Sphingosine-1-phosphate Receptor Subtype-1 Agonists. Bioorg. Med. Chem. Lett. 2010, 20, 2520–2524. [DOI] [PubMed] [Google Scholar]
- Evindar G.; Deng H.; Bernier S. G.; Doyle E.; Lorusso J.; Morgan B. A.; Westlin W. F. Exploring Amino Acids Derivatives As Potent, Selective, and Direct Agonists of Sphingosine-1-phosphate Receptor Subtype-1. Bioorg. Med. Chem. Lett. 2013, 23, 472–475. [DOI] [PubMed] [Google Scholar]
- Albert R.; Hinterding K.; Brinkmann V.; Guerini D.; Hartwieg C.-M.; Knecht H.; Simeon C.; Streiff M.; Wagner T.; Welzenbach K.; Zecri F.; Zollinger M.; Cooke N.; Francotte E. Novel Immunomodulator FTY720 Is Phosphorylated in Rats and Humans to Form a Single Stereoisomer. Identification, Chemical Proof, and Biological Characterization of the Biologically Active Species and Its Enantiomer. J. Med. Chem. 2005, 48, 5373–5377. [DOI] [PubMed] [Google Scholar]
- Fyrst H.; Saba J. D. An Update on Sphingosine-1-phosphate and Other Sphingolipid Mediators. Nat. Chem. Biol. 2010, 6, 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billich A.; Bornancin F.; Dévay P.; Mechtcheriakova D.; Urtz N.; Baumruker T. Phosphorylation of the Immunomodulatory Drug FTY720 by Sphingosine Kinases. J. Biol. Chem. 2003, 278, 47408–47415. [DOI] [PubMed] [Google Scholar]
- Davis D. D; Clemens J. J.; Macdonald T. L.; Lynch R. K. Sphingosine-1-phosphate Analogs as Receptor Antagonists. J. Biol. Chem. 2005, 280, 9833–9841. [DOI] [PubMed] [Google Scholar]
- Constantinescu C. S.; Farooqi N.; O’Brien K.; Gran B. Experimental Autoimmune Encephalomyelitis (EAE) as a Model for Multiple Sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik J. M.; Schmouder R. L.; Slade A. J. Overview of FTY720 Clinical Pharmacokinetics and Pharmacology. Ther. Drug Monit. 2004, 26, 585–587. [DOI] [PubMed] [Google Scholar]
- Kovarik J. M.; Schmouder R.; Barilla D.; Wang Y.; Kraus G. Single-Dose FTY720 Pharmacokinetics, Food Effect, and Pharmacological Responses in Healthy Subjects. Br. J. Clin. Pharmacol. 2004, 57, 586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anson M. S.; Graham J. P; Roberts A. J. Development of a Fully Telescoped Synthesis of the S1P1 Agonist GSK1842799. Org. Process Res. Dev. 2011, 15, 649–659. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.