

Abstract

The bacterial RNA polymerase (RNAP) is an ideal target for the development of antimicrobial agents against drug-resistant bacteria. Especially, the switch region within RNAP has been considered as an attractive binding site for drug discovery. Here, we designed and synthesized a series of novel hybrid-type inhibitors of bacterial RNAP. The antimicrobial activities were evaluated using a paper disk diffusion assay, and selected derivatives were tested to determine their MIC values. The hybrid-type antimicrobial agent 29 showed inhibitory activity against Escherichia coli RNAP. The molecular docking study suggested that the RNAP switch region would be the binding site of 29.
Keywords: bacterial RNA polymerase, switch region, antimicrobial agent, bacterial RNA polymerase inhibitor, hybrid
Rifamycins are a class of bacterial RNA polymerase (RNAP) inhibitors commonly used in clinical practice. The potency and broad-spectrum nature of rifamycins have improved the treatment of infectious diseases, including tuberculosis. Nevertheless, the emergence of drug-resistant bacteria poses an ongoing threat to the therapeutic index of rifamycins. For this reason, the development of a novel RNAP inhibitor is in demand. In 2008, a research team led by Arnold and Ebright elucidated the inhibitory mechanism of myxopyronin A (1) (Figure 1),1 which was targeting the switch region of bacterial RNAP, and an X-ray crystal structure of 1 in complex with T. thermophilus RNAP was reported.2,2b Corallopyronin (3) and ripostatins also appeared to bind to the RNAP switch region.2a Hence, this newly identified binding site in RNAP has attracted the attention of medicinal chemists to develop new antimicrobial agents.
Figure 1.

Structures of antibiotics myxopyronins A (1) and B (2), corallopyronin (3), and holomycin (4).
Before now, the research group of Simonsen and Xiang attempted a direct structure–activity relationship study of myxopyronin.3,3b In addition, Fishwick and co-workers tried structure-based ligand design at the myxopyronin binding site.4 On the other hand, Moy and co-workers reported that myxopyronin was an unsuitable lead compound for medicinal development because of the high binding to serum albumin, the low stability, and the occurrence of resistant strains.5
In this report, we disclose our effort to develop novel antimicrobial agents with a hybrid strategy6,6b for myxopyronin derivatives. To enhance the antimicrobial activity, reduce the lipophilicity, and improve the instability, the antibiotic holomycin (4)7 was incorporated into the mother skeleton of the myxopyronin-type RNAP inhibitors.
Holomycin (4) also possessed antibacterial properties and provided moderate but broad antibiotic spectrum against Gram-positive and -negative bacteria.8,8b In addition, 4 showed the inhibition of RNA synthesis,8a and the calculate log P (ClogP) did not exceed 2.0.9 These properties fascinated us to hybridize myxopyronins with the holothin molecule to expect the efficacy beyond our anticipations.
Our hybrid strategy is shown in Figure 2. We designed two types of hybrid derivatives based on the reported X-ray crystal structure of RNAP with 1 (PDB: 3DXJ).2,2b One was the “right-hand amide derivative (6)” with the core α-pyrone at the center and the holothin component positioned on the right-hand side. Toward the right side of the X-ray crystal structure, as shown in the blue circle in Figure 2, there is an intermolecular hydrogen bond network involving a water molecule. The holothin (5), which included several heteroatoms, was incorporated into the right-hand moiety to form the hydrogen bonds. The other is the “left-hand amide derivative (7)”. The holothin was introduced at the left side of the molecule relative to the C3 side chain. At the left side of the myxopyronin binding site, as shown in the red circle in Figure 2, there is a useful space around the C20 hydrocarbon chain. This interspace was advantageous for accommodating the holothin moiety to increase the interaction with RNAP.
Figure 2.
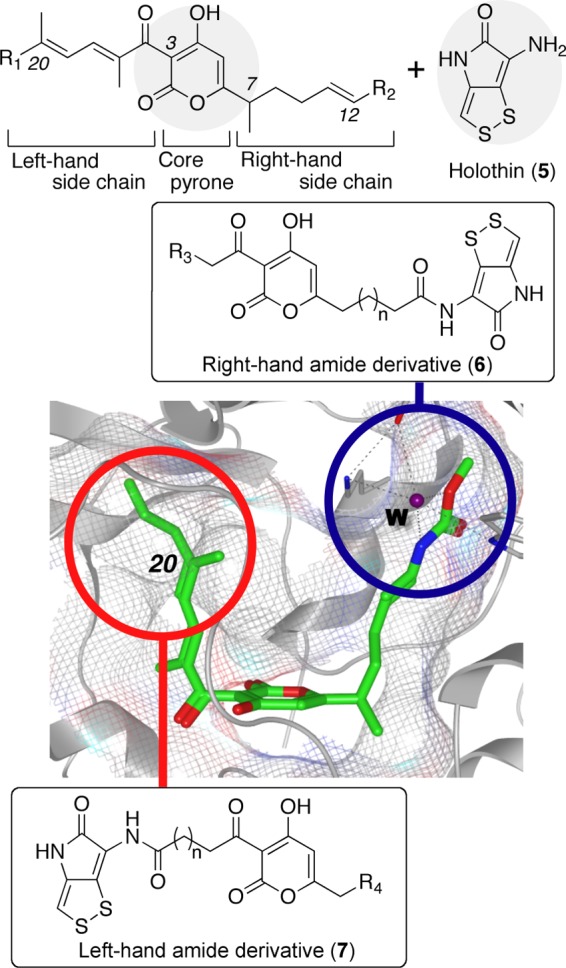
Design of hybrid-type derivatives including core α-pyrone and holothin (5) based on the X-ray crystallography of 1 within bacterial RNAP from T. thermophilus (PDB: 3DXJ; green stick, myxopyronin A; gray stick and ribbon, bacterial RNAP; gray mesh, myxopyronin binding site; W, water molecule; blue circle, intermolecular hydrogen bond network at the right-hand side; and red circle, space at the left-hand side).
We first synthesized right-hand amide derivatives (Scheme 1). The C3-acylation of starting material 8 proceeded in the presence of propionic acid via condensation and rearrangement to obtain 9.10 The pyrone 9 was alkylated with 10,11 then the silyl protecting group was removed, and two-step oxidation produced the carboxylic acid 11.3a Compound 11 was treated with oxalyl chloride and condensed with holothin hydrochloride 12(12) under basic conditions to yield the simplified hybrid derivative 13. On the other hand, 17 was synthesized to ascertain the importance of the left-hand portion of the hybrid compound. Compound 8 was acylated with nonanoic acid, and the resultant pyrone 14 was converted to a carboxylic acid 16 through the same four steps. Compound 16 was condensed with the holothin salt 12 to construct the desired 17. We attempted to prepare a series of right-hand derivatives with unsaturated carbon side chains; however, the final products could not be isolated due to decomposition at the purification step. These observations suggested that the unsaturated side chain of 1 was one factor for the reported instability.5
Scheme 1. Syntheses of the Right-Hand Amide Derivatives 13 and 17.

Reagents and conditions: (a) Propionic acid or nonanoic acid, DIC, DMAP, toluene, 100 °C, 5 or 14 h, 72 or 74%, respectively. (b) Compound 10 or 15, LDA, HMPA, THF, −78 °C, 1 h, 75%. (c) AcOH/THF/H2O (3/1/1), rt, 26 h, 66–89%. (d) PCC, CH2Cl2, rt, 30 min. (e) NaClO2, NaH2PO4, 2-methyl-2-butene, tBuOH/THF/H2O (1/1/1), rt, 4 h, 63–84%. (f) (COCl)2, toluene, rt, 1 h. (g) Compound 12, Et3N, THF/toluene, rt, 30 min, 61–65%.
We next attempted to synthesize the left-hand amide derivatives. As shown in Scheme 2A, 8 and different lengths of the hydrocarbon linkers 18–20 were treated with N,N′-diisopropylcarbodiimide (DIC) and DMAP for the C3-acylation.10 The methyl ester groups of the obtained 21–23 were then hydrolyzed to the carboxylic acids 24–26, followed by the condensation of 24–26 with 12 to generate 27–29. Compound 28 was subsequently converted to the sulfone 30 using oxone as the oxidizing reagent.13,13b
Scheme 2. Syntheses of the Left-Hand Amide Derivatives 27–29, 30, 32, and 35.

Reagents and conditions: (a) Compound 18, 19, or 20, DIC, DMAP, toluene, 100 °C, 6–14 h, 64–93%. (b) 1 M LiOH aq/THF (1/4), rt, 14 h. (c) (COCl)2, toluene, rt, 1 h. (d) Compound 12, Et3N, THF/toluene, rt, 30 min, 38–66%. (e) Oxone, acetone/H2O, 0 °C, 30 min, 40%. (f) 4-Bromo-1-butene, LDA, HMPA, THF, rt, 14 h, 85%. (g) Ba(OH)2, H2O/THF, 73 °C, 2 h.
The synthetic route B (Scheme 2B) showed the synthesis of the derivative 32 including the right-hand alkyl chain and the left-hand amide part. Compound 8 was alkylated using 4-bromo-1-butene, and the C3 position of the obtained pyrone was acylated with 19. After hydrolysis, the resultant carboxylic acid 31 was condensed with 12 to obtain 32.
The synthetic route C (Scheme 2C) describes the preparation of a C3 amide derivative. The amide ester analogue 33, prepared from 4-hydroxy-6-methyl-2-oxo-2H-pyran-3-carboxylic acid14 and methyl 7-aminoheptanoate, was mildly hydrolyzed to 34, and then, the carboxylic acid was condensed with 12 to synthesize the product 35.
As other derivatives, three compounds 36–38 were also synthesized (Figure 3).15 Compound 36 was constructed to ascertain the significance of the length of the left-hand hydrocarbon linker. The piperidine-containing derivative 37 was synthesized to examine the effect of the holothin component. The holothin derivative 38(16) was also synthesized to observe the efficacy of a hybrid structure including α-pyrone.
Figure 3.

Structures of the synthesized derivatives 36–38.
The synthesized derivatives were evaluated as an antimicrobial agent using a simple paper disk diffusion assay against Bacillus subtilis (Table 1).17 The right-hand amide derivatives were tested first. The synthetic intermediate 9 did not exhibit a zone of inhibition. The right-hand amide derivative 13 also showed no antimicrobial activity. By contrast, 14, 16, and 17, which included left-hand side chains, produced inhibition zones. These results suggested that the left-hand side chain played an important role in the antimicrobial activity. Furthermore, it became clear that the antimicrobial properties of this series did not depend on the holothin moiety.
Table 1. Disk Diffusion Assay against B. subtilis.
| compda | inhibition zoneb (mm) | compda | inhibition zoneb (mm) |
|---|---|---|---|
| 9 | no zone | 31 | no zone |
| 13 | no zone | 32 | 10 |
| 14 | 12 | 34 | no zone |
| 16 | slight | 35 | slight |
| 17 | 9 | 36 | 9 |
| 25 | no zone | 37 | no zone |
| 27 | 14 | 38 | 9 |
| 28 | 11 | RFP | 25 |
| 29 | 15 | (±)-2 | slight |
| 30 | no zone | MeOH | no zone |
All compounds were dissolved in MeOH and evaluated at a dose of 30 μg/disk.
Diameter of zones with complete growth inhibition.
Next, left-hand amide derivatives were evaluated. As compared with the carboxylic acid 25 and the hybrid derivatives 27–29, the hybrid-type derivatives showed zones of inhibition. These results suggested that the presence of the holothin moiety enhanced the activity. Compound 30, including a sulfone group, and 31 and 32 with right-hand hydrocarbon chains were compared with 28; however, these derivatives did not show significant validity, suggesting that the sulfone group and the right-hand chains did not contribute to the activity. Furthermore, 34 did not show any activity, although 35 exhibited a slight zone of inhibition. The α-pyrone 36 and the holothin derivative 38 showed weak activities, whereas 37 produced no zone of inhibition. The synthesized (±)-myxopyronin B11 was also tested; however, (±)-2 produced only a slight zone of inhibition in our assay system. Therefore, the authentic rifampicin (RFP) was used as a standard antibiotic in all assays.
The results in Table 1 indicated that the C3 position of the α-pyrone should be substituted with a long hydrocarbon moiety for the expression of the antimicrobial activity. In addition, the hybrid derivatives containing α-pyrone and the holothin moiety exhibited the enhanced activity.
On the basis of results of the paper disk assay, we selected the effective derivatives 14, 27, and 29 for the next evaluations of higher order assessments. These simplified derivatives presented larger zones of inhibition, small molecular weights, and low lipophilicities, as assessed by ClogP.9
The minimal inhibitory concentration (MIC) was evaluated to characterize the antimicrobial activity of 14, 27, and 29 against eight kinds of Gram-positive and -negative bacteria and fungi (Table 2). After 24 h of incubation, α-pyrone-type derivative 14(18) and the hybrid-type derivative 29 showed good antimicrobial activity against Gram-positive bacteria, although the activity against Gram-negative bacteria and fungi was weak. The extension of the left-hand chain was effective for the inhibition against the Gram-positive bacteria. Furthermore, 14, 27, and 29 gave the strongest activity against Micrococcus luteus. The selective inhibitory activity against bacteria would depend on our hybrid derivatives.
Table 2. MIC Values for Selected Derivatives 14, 27, and 29.
| MIC (μg/mL) |
||||
|---|---|---|---|---|
| organism | 14 | 27 | 29 | RFP |
| Staphylococcus aureus ATCC29213 | 8 | 64 | 4 | 0.5 |
| Enterococcus faecalis ATCC29212 | 4 | 16 | 4 | 1 |
| M. luteus ATCC9341 | 2 | 8 | 1 | ≤0.125 |
| B. subtilis ATCC6633 | 8 | 64 | 16 | ≤0.125 |
| E. coli ATCC25922 | 128 | 128 | ≥256 | 8 |
| Pseudomonas aeruginosa ATCC27853 | 128 | 64 | 128 | 32 |
| Serratia marcescens ATCC13880 | 128 | 128 | ≥256 | 32 |
| Candida albicans ATCC10231 | 64 | 128 | ≥256 | ≥256 |
Besides, 14, 27, and 29 were evaluated for their in vitro inhibitory activity against Escherichia coli. RNAP (Table 3). The derivative 29 inhibited E. coli RNAP, and the IC50 value was determined to be 14 ± 1.7 μM. Unfortunately, 14 and 27 did not exhibit inhibitory activity. The IC50 value of 29 was higher than that of RFP, but the difference of activities might be a result of the binding site recognition.2b In addition, the ineffectiveness of 29 against E. coli in MIC might be due to the low penetration ability in Gram-negative bacteria. However, the results in Table 3 suggested that the target of 29 was the bacterial RNAP.
Table 3. In Vitro Inhibitory Activity against E. coli RNA Polymerase.
From the above, the expected binding site of 29 in RNAP was considered in a molecular docking study based on X-ray crystal data of myxopyronin A and RNAP complex (PDB: 3DXJ).19 As shown in Figure 4, 29 showed good conformation in the hydrophobic pocket of the switch region with a minimum binding energy of −8.3 kcal/mol. Other predicted conformations of 29 within RNAP were also examined; however, these provided high binding energies over 13 kcal/mol at least. Consequently, the expected binding site of 29 within RNAP would be regarded as the switch region.
Figure 4.
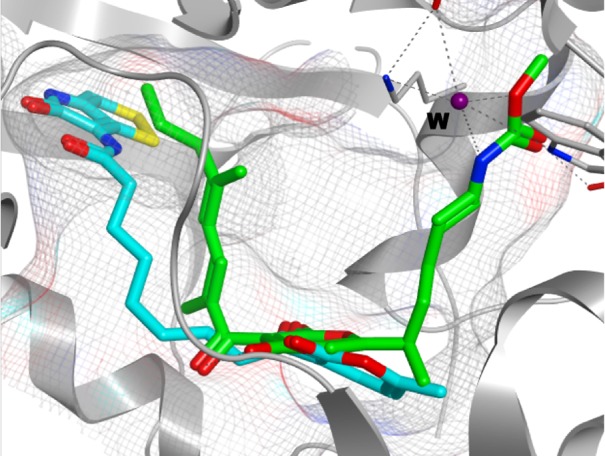
Molecular docking study of 29 in the switch region of T. thermophilus RNAP (PDB: 3DXJ). Binding energy, −8.3 kcal/mol (blue stick, 29; green stick, myxopyronin A; gray stick and ribbon, bacterial RNAP; gray mesh, myxopyronin binding site; and W, water molecule).
In summary, we designed and synthesized hybrid-type antimicrobial derivatives including the core α-pyrone and the holothin moiety toward the development of novel bacterial RNAP inhibitors. The simplified derivative 29, in particular, showed reliable antimicrobial activity against Gram-positive bacteria. Furthermore, 29 inhibited E. coli RNAP, and the molecular docking study suggested that the targeted switch region would be the binding site. Our hybrid strategy revealed that the left-hand moiety of the α-pyrone component had a great effect on the antimicrobial activity, and the inserted holothin part enhanced the ability. In addition, a derivative with low lipophilicity and inhibitory activity against bacterial RNAP was identified. Significantly, selectivity of antimicrobial action and profiles of our hybrid-type derivatives indicated the unexpected validity of hybridization. Hence, the hybrid strategy would be one of the effective ways to explore novel drug candidates. The structure–activity relationship study of the simplified derivatives and the X-ray crystallographic analysis of the cocrystal (29 and bacterial RNAP) are in progress.
Acknowledgments
We thank C. Sakuma of Tokyo University of Pharmacy and Life Sciences for NMR and mass spectroscopy measurements.
Supporting Information Available
General information, details of the compound syntheses and characterization, and the biological assay protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
This research was supported by grants from MEXT (the Ministry of Education, Culture, Sports, Science and Technology), Japan, including the Platform for Drug Discovery, Informatics, and Structural Life Science, the Grant-in-Aid for Research Activity Start-up (No. 21890262), and the Grant-in-Aid for Young Scientists (B) (No. 23790142).
The authors declare no competing financial interest.
Supplementary Material
References
- Irschik H.; Gerth K.; Höfle G.; Kohl W.; Reichenbach H. The myxopyronins, new inhibitors of bacterial RNA synthesis from Myxococcus fulvus (Myxobacterales). J. Antibiot. 1983, 36, 1651–1658. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay J.; Das K.; Ismail S.; Koppstein D.; Jang M.; Hudson B.; Sarafianos S.; Tuske S.; Patel J.; Jansen R.; Irschik H.; Arnold E.; Ebright R. H. The RNA polymerase “switch region” is a target for inhibitors. Cell 2008, 135, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M. X.; Hudson B. P.; Das K.; Arnold E.; Ebright R. H. Structures of RNA polymerase-antibiotic complexes. Curr. Opin. Struct. Biol. 2009, 19, 715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doundoulakis T.; Xiang A. X.; Lira R.; Agrios K. A.; Webber S. E.; Sisson W.; Aust R. M.; Shah A. M.; Showalter R. E.; Appleman J. R.; Simonsen K. B. Myxopyronin B analogs as inhibitors of RNA polymerase, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2004, 14, 5667–5672. [DOI] [PubMed] [Google Scholar]
- Lira R.; Xiang A. X.; Doundoulakis T.; Biller W. T.; Agrios K. A.; Simonsen K. B.; Webber S. E.; Sisson W.; Aust R. M.; Shah A. M.; Showalter R. E.; Banh V. N.; Steffy K. R.; Appleman J. R. Syntheses of novel myxopyronin B analogs as potential inhibitors of bacterial RNA polymerase. Bioorg. Med. Chem. Lett. 2007, 17, 6797–6800. [DOI] [PubMed] [Google Scholar]
- McPhillie M. J.; Trowbridge R.; Mariner K. R.; O’Neill A. J.; Johnson A. P.; Chopra I.; Fishwick C. W. G. Structure-based ligand design of novel bacterial RNA polymerase inhibitors. ACS Med. Chem. Lett. 2011, 2, 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy T. I.; Daniel A.; Hardy C.; Jackson A.; Rehrauer O.; Hwang Y. S.; Zou D.; Nguyen K.; Silverman J. A.; Li Q.; Murphy C. Evaluating the activity of the RNA polymerase inhibitor myxopyronin B against Staphylococcus aureus. FEMS Microbiol. Lett. 2011, 319, 176–179. [DOI] [PubMed] [Google Scholar]
- See the review:; Tietze L. F.; Bell H. P.; Chandrasekhar S. Natural product hybrids as new leads for drug discovery. Angew. Chem., Int. Ed. 2003, 42, 3996–4028. [DOI] [PubMed] [Google Scholar]
- Tsogoeva S. B. Recent progress in the development of synthetic hybrids of natural or unnatural bioactive compounds for medicinal chemistry. Mini Rev. Med. Chem. 2010, 10, 773–793. [DOI] [PubMed] [Google Scholar]
- Kenig M.; Reading C. Holomycin and an antibiotic (MM19290) related to tunicamycin, metabolites of Streptomyces clavuligerus. J. Antibiot. 1979, 32, 549–554. [DOI] [PubMed] [Google Scholar]
- Oliva B.; O’Neill A.; Wilson J. M.; O’Hanlon P. J.; Chopra I. Antimicrobial properties and mode of action of the pyrrothine holomycin. Antimicrob. Agents Chemother. 2001, 45, 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill A.; Oliva B.; Storey C.; Hoyle A.; Fishwick C.; Chopra I. RNA polymerase inhibitors with activity against rifampin-resistant mutants of Staphylococcus aureus. Antimicrob. Agents Chemother. 2000, 44, 3163–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The value of calculate Log P (ClogP) was calculated by CS ChemBioDraw Ultra 12.0 (Cambridgesoft.com).
- Mori T.; Ujihara K.; Matsumoto O.; Yanagi K.; Matsuo N. Synthetic studies of fluorine-containing compounds for household insecticides. J. Fluorine Chem. 2007, 128, 1174–1181. [Google Scholar]
- Hu T.; Schaus J. V.; Lam K.; Palfreyman M. G.; Wuonola M.; Gustafson G.; Panek J. S. Total synthesis and preliminary antibacterial evaluation of the RNA polymerase inhibitors (±)-myxopyronin A and B. J. Org. Chem. 1998, 63, 2401–2406. [Google Scholar]
- Compound 12 was synthesized according to the following manuscript.Hjelmgaard T.; Givskov M.; Nielsen J. Expedient total synthesis of pyrrothine natural products and analogs. Org. Biomol. Chem. 2007, 5, 344–348. [DOI] [PubMed] [Google Scholar]
- Adam W.; Chan Y.-Y.; Cremer D.; Gauss J.; Scheutzow D.; Schindler M. Spectral and chemical properties of dimethyldioxirane as determined by experiment and ab initio calculations. J. Org. Chem. 1987, 52, 2800–2803. [Google Scholar]
- Yu B.; Liu A.-H.; He L.-N.; Li B.; Diao Z.-F.; Li Y.-N. Catalyst-free approach for solvent-dependent selective oxidation of organic sulfides with oxone. Green Chem. 2012, 14, 957–962. [Google Scholar]
- Suzuki E.; Sekizaki H.; Inoue S. A new and simple synthesis of triacetic acid lactone-3-carboxylic acid (3-carboxy-4-hydroxy-6-methyl-2-pyrone). Synthesis 1975, 652–653. [Google Scholar]
- See the Supporting Information.
- Shiozawa H.; Kagasaki T.; Kinoshita T.; Haruyama H.; Domon H.; Utsui Y.; Kodama K.; Takahashi S. Thiomarinol, a new hybrid antimicrobial antibiotic produced by a marine bacterium fermentation, isolation, structure, and antimicrobial activity. J. Antibiot. 1993, 46, 1834–1842. [DOI] [PubMed] [Google Scholar]
- National Committee for Laboratory Standards. Performance Standards for Antimicrobial Disk Susceptibility Tests; Approved standard M2-A8; NCCLS: Wayne, 2003. [Google Scholar]
- The similar derivative of 14, 4-hydroxy-6-methyl-3-octanoyl-2H-pyran-2-one, was reported as an antibacterial agent.Ukita T.; Mizuno D.; Tamura T.; Yamakawa T.; Nojima S. The antibacterial properties of compounds containing the tricarbonylmethane group. II-III. Antibacterial properties of 3-alkylated or 3-acylated 4-hydroxycoumarins and of a related pyrone. J. Pharm. Soc. Jpn. 1951, 71, 234–243. [Google Scholar]
- The molecular docking study was performed using the molecular modeling package MOE 2010.10 (Chemical Computing Group, Inc., Montreal, Canada: ). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.