

Abstract
The World Health Organization has classified the leishmaniasis as a major tropical disease. The discovery of new compounds for leishmaniasis is therefore a pressing concern for the anti-infective research program. We have synthesized 19 compounds of triazine dimers as novel antileishmanial agents. Most of the synthesized derivatives exhibited better activity against intracellular amastigotes (IC50 ranging from 0.77 to 10.32 μM) than the control, pentamidine (IC50 = 13.68 μM), and are not toxic to Vero cells. Compounds 14 and 15 showed significant in vivo inhibition of 74.41% and 62.64%, respectively, in L. donovani/hamster model. Moreover, expansion of Th1-type and suppression of Th2-type immune responses proved that compound 14 stimulates mouse macrophages to prevent the progression of leishmania parasite. The molecular docking studies involving PTR1 protein PDB further validated the concepts involved in the design of these compounds. Among the investigated analogues, compound 14 has emerged as the potential one to enlarge the scope of the study.
Keywords: L. donovani, triazine, pentamidine, in vitro/in vivo, pteridine reductase 1
Leishmaniasis is a group of tropical disease resulting from infection of macrophage by obligate intracellular parasites of genus Leishmania, and it is a major health problem worldwide.1 It may manifest in three clinical forms, cutaneous, muco-cutaneous, and visceral leishmaniasis (VL). Among all, VL is the most severe form of the disease.2 Out of the annual 2 million cases reported every year, 500 000 are due to VL thus causing a significant health problem.3Leishmania infection is classically associated with a depression of T helper type 1 (Th1) cells and preferential expansion of T helper type 2 (Th2) cells. Th1 type immune responses play an important role in mediating protection against Leishmania,4 including roles of interferon (IFN)-γ, interleukin (IL)-12, tumor necrosis factor (TNF)-α, and nitric oxide (NO), whereas inhibitory effects have been reported for Th2 cytokines, IL-10, and transforming growth factor (TGF)-β.5,6 Control of Leishmania infection depends on IL-12-driven expansion of Th1 cells, macrophage activation through production of IFN-γ, and the subsequent generation of nitric oxide (NO).7 The serious problems associated with the treatment of leishmania infections, such as expensive and toxic drugs, emerging resistance, and the limited funding, have recently led many research groups to design and synthesize safer, inexpensive, and more efficient novel antileishmanial compounds.
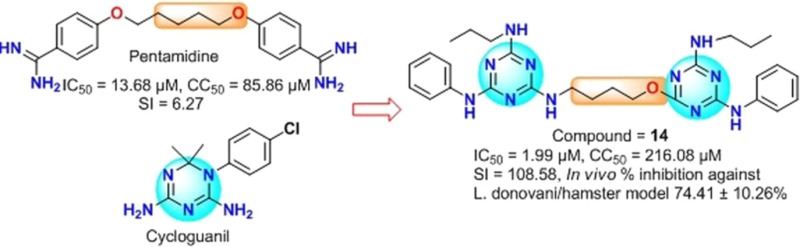
In this regard, pentamidine8 has been used to treat early stage Trypanosoma brucei gambiense related Human African Trypanosomiasis,9,10 and antimony resistant VL, which was caused by Leishmania donovani.11,12 Because of fewer side effects and low propensity to the development of resistance, pentamidine may be a viable starting point for the development of novel antileishmanial agents.13−15 On the other side, triazine scaffold provides the basis for the design of biologically relevant molecules with extensive application as therapeutics. Previously, it was found that triazine based scaffolds showed various biological activity such as antimalarial, antileishmanial, and antimicrobial activity.16 We have recently gained interest in the synthesis of pentamidine, pyrimidine, triazine, and quinazolinone derivatives that showed a marked antileishmanial activity.17−19
Dihydrofolate reductase (DHFR) is a well established drug target for a wide range of diseases.20 This pathway has been effectively used against cancer, bacterial infections, and malaria, but with limited success against Leishmaniasis.21 Interestingly, Leishmania is known to proficiently overcome DHFR inhibition by overexpressing pteridine reductase 1 (PTR1, EC 1.5.1.33). Pteridine reductase (PTR) is associated with the pterin and folate metabolism and essential for the growth of Leishmania. Methotrexate and triaminoquinazolines are some known potential inhibitors of this protozoan enzyme.22 Hence, the PTR1 pathway may offer unique opportunity to develop new chemotherapeutics, which could be effective against Leishmania.23,24 As part of our ongoing anti-infective research program,25,26 considering the structural similarity between the key structural units of PTR1 inhibitors (pteridines and quinazolines) and benzimidamide moiety of pentamidine (a well-known antileishmanial agent), we have synthesized the triazine dimers and evaluated them for their antileishmanial activity. In view of aforesaid, we have also carried out docking studies of these compounds to investigate their plausible mode of binding and affinity to the PTR1.
We have used cyanuric chloride, an inexpensive commercially available reagent as the starting material for the synthesis of compounds (5–23). Herein, we report a synthetic procedure for triazine compounds (5–23), which is depicted in Scheme 1. Trisubstituted triazines derivatives were synthesized; starting from the treatment of aniline 1 with cyanuric chloride at 0 °C to give monosubstituted triazine derivatives 2 in excellent yield (>95%). In the next step, compound 2 reacted with aminoalkanol at room temperature to give compounds 3 and 4 in good yield. Further, the compounds 3 and 4 with 2 were reacted in the presence of sodium hydride in dry THF to afford the heterodimeric intermediates 5 and 6 in moderate yield (∼50%). Thus, isolated compound 5 and 6 were treated with different amines at 60 °C to give the final products in high yield (80–95%).
Scheme 1. Synthesis of Triazine Mimetics (5–23).

Reagents and conditions: (a) cyanuric chloride, 0 °C; (b) aminoalkanol, K2CO3, THF, rt; (c) monosubstituted triazine; (2), NaH, THF, rt; (d) RNH2, THF, 60 °C.
In this endeavor to identify the activity of triazines, we have synthesized a series of 19 compounds and screened them against L. donovani. The results are summarized in Table 1.
Table 1. In Vitro Antileishmanial Activity and Cytotoxicity of Triazine Mimetics (5–23)a.
| entry | antiamastigote activity IC50b (μM) | cytotoxicity CC50c (μM) | selectivity indexd (SI) |
|---|---|---|---|
| 5 | 8.98 ± 0.73 | 17.10 ± 2.61 | 1.90 |
| 6 | 10.32 ± 1.87 | 7.74 ± 1.2 | 0.75 |
| 7 | >40 | NDe | NAf |
| 8 | >40 | ND | NA |
| 9 | 3.15 ± 0.80 | 275.63 ± 11.78 | 87.50 |
| 10 | 2.91 ± 0.32 | 115.27 ± 5.98 | 39.61 |
| 11 | 1.70 ± 0.54 | 13.80 ± 2.14 | 8.11 |
| 12 | 6.46 ± 1.20 | >400 | >61.92 |
| 13 | 3.21 ± 0.67 | >400 | >124.61 |
| 14 | 1.99 ± 0.31 | 216.08 ± 5.89 | 108.58 |
| 15 | 0.77 ± 0.15 | 198.26 ± 9.98 | 257.48 |
| 16 | 3.51 ± 0.81 | >400 | >113.96 |
| 17 | >40 | ND | NA |
| 18 | 1.33 ± 0.51 | 20.06 ± 2.52 | 15.08 |
| 19 | 0.86 ± 0.06 | 16.23 ± 2.27 | 18.87 |
| 20 | 8.98 ± 0.98 | 17.10 ± 1.10 | 1.90 |
| 21 | 7.52 ± 1.19 | >400 | >53.19 |
| 22 | 2.65 ± 0.54 | >400 | >150.94 |
| 23 | 8.26 ± 0.62 | 18.23 ± 4.37 | 2.20 |
| SSGg | 46.70 ± 2.75 | >400 | 8.56 |
| pentamidine | 13.68 ± 1.57 | 85.86 ± 5.27 | 6.27 |
| miltefosine | 8.10 ± 0.70 | 53.12 ± 4.75 | 6.56 |
IC50 and CC50 values are the average of two independent assays expressed as average ± standard deviation.
IC50 on L. donovani intracellular amastigotes.
CC50 on Vero cells.
The selectivity index (SI) is defined as the ratio of CC50 to IC50.
ND: not determined.
NA: not available.
SSG: Sodium stibogluconate.
Initially, we have synthesized the triazine derivatives (5, 7, and 8) with 5-aminopentan-1-ol linker, considering the importance of standard drug pentamidine, but unfortunately, these compounds were found to be inactive against Leishmania donovani. Further to improve the activity of these compounds, we have synthesized the triazine analogues with shorter chain length. Interestingly, all the synthesized derivatives showed significant to moderate antiamastigote activity with IC50 ranging from 0.77 to 10.32 μM. Most of the synthesized compounds having 4-aminobutan-1-ol linker responded better than the control drugs in the test system and displayed significant activity. First line drug, sodium stibogluconate (SSG) displayed a poor in vitro activity against Leishmania amastigotes (IC50 = 46.70 μM), although it is not toxic to Vero cells (CC50 > 400 μM). Other standard drugs, pentamidine and miltefosine displayed moderate in vitro antiamastigote activity (IC50 = 13.6 and 8.10 μM, respectively) and low cytotoxicity toward Vero cells (CC50 values of 85.86 and 53.12 μM). Our synthesized intermediate 6, which has a chloro substituent at R1 and R2, exhibited moderate activity (IC50 = 10.32 μM) and cytotoxicity toward Vero cells (CC50 = 7.74 μM). In compound 6, when we replaced the chloro with morpholine (9) (IC50 = 3.15 μM and CC50 = 275.6 μM), amino ethyl morpholine (10) (IC50 = 2.91 μM and CC50 = 115.27 μM), and amino propyl morpholine (11) (IC50 = 1.70 μM and CC50 = 13.80 μM), the resulting compounds showed successive increase in the antileishmanial activity with concomitant reduction in cytotoxicity. For the structure–activity relationship (SAR), we have introduced the aliphatic amines at R1 and R2 such as n-pentyl amine containing analogues (12), which displayed good antiamastigote activity (IC50 = 6.46 μM) and better selectivity (>61.92). However, decreasing the chain length by one carbon (R1 = R2 = n-butyl) enhanced the activity of compound 13 (IC50 = 3.21 μM), and interestingly, further decrease in chain length by one carbon (R1 = R2 = n-propyl, 14) demonstrated further enhancement in antiamastigote activity (IC50 = 1.99 μM) and selectivity (108.58). Surprisingly, tert-butyl substituted derivative 15 exhibited better activity (IC50 = 0.77 μM) and selectivity (257.48) than iso-propyl substituted derivative 16 (IC50 = 3.51 μM; SI > 113.96) and was found best among all the synthesized analogues. Notably, all the n-amino alkyl substituted derivatives were found to be nontoxic to Vero cells (CC50 ranging from 198.26 to 400 μM). Unexpectedly, in the case of n-cyclopropyl substituted compound 17, no significant activity was obtained. Compounds 18 and 19, in which chlorine atom were replaced by N-methylpiperazine and N-ethylpiperazine showed excellent activity with IC50 = 1.33 and 0.86 μM and CC50 = 20.06 and 16.23 μM, respectively. Additionally, tetrahydroquinoline containing analogue 20 exhibited moderate antiamastigote activity (IC50 = 8.98 μM) and poor selectivity (>1.90), whereas the tetrahydroisoquinoline containing analogue 21 displayed comparable antiamastigote activity (IC50 = 7.52 μM) and better selectivity (SI > 53.19) than 20 representing that the position of nitrogen atom in the tetrahydro ring is crucial for selectivity. The above observations tempted us to check the effect of n-cyclopropyl on SAR. For this purpose, we have synthesized compounds 22 and 23, which bear different alkyl amine at R1 and R2. Interestingly, compound 22 (R1 = n-propyl, R2 = n-cyclopropyl) was found to be effective (IC50 = 2.65 μM) with good selectivity (SI = >150.94), and compound 23 (R1 = n-cyclopropyl and R2 = n-propyl) showed significant but lesser potency (IC50 = 8.26 μM) and poor selectivity (2.20) than compound 22.
On the basis of excellent in vitro potency and selectivity index, we have selected compounds 14 and 15 as potential candidates for further evaluation; accordingly, in vivo tests were performed in the L. donovani/golden hamster model. The aqueous suspensions of test compounds were administered for five consecutive days at 50 mg/kg/day by intraperitoneal (IP) route. The post-treatment (p.t.) splenic biopsies were done on day 7 of the last dose administration and amastigote counts were assessed by Giemsa staining. Compound 14 showed good percentage inhibition (74.41 ± 10.26%) in parasite multiplication, and compound 15 has shown moderate in vivo efficacy (62.64 ± 10.74%). Furthermore, we have not observed any abnormal behavior of animals at the time of dosing (Figure 1).
Figure 1.
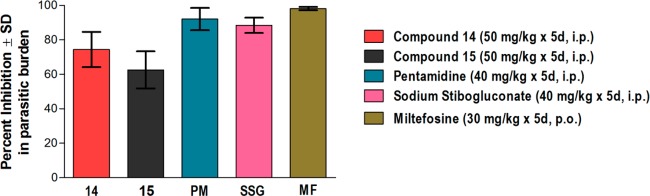
In vivo antileishmanial efficacy in L. donovani/golden hamster model. Each bar represents pooled data (mean ± SD value) of two experiments. Five animals were used in each experiment. Pentamidine (PM), sodium stibogluconate (SSG), and miltefosine (MF) are used as reference drugs. SD, standard deviation; i.p., intraperitoneal; p.o., per oral.
Potent in vitro and in vivo efficacy of compound 14 prompted us to explore its immunostimulatory properties whether it is able to reverse immunosuppression in Leishmania infected mouse macrophages (J-774A.1). Our investigations clearly indicate that compound 14 treated cells induce the Th1 type immune responses by remarkable production of IL-12, TNF-α, and nitric oxide (NO) and effective suppression of Th2 type cytokines, IL-10 and TGF-β (Figure 2). We have observed significant (p < 0.05) increase in the production of IL-12 (3-fold more than infected control), TNF-α (2-fold more than infected control), and NO (p < 0.01, 2-fold higher than untreated infected macrophages) at 24 h post-treatment (p.t.). In contrast, among the Th2 type cytokines, there was considerable (p < 0.05) suppression of IL-10 observed at 24 h p.t. The level of TGF-β was also down-regulated in compound 14 treated cells (1.8-fold less production than infected cells). Our study also revealed that after 24 h p.t., all the cytokines and nitric oxide started attaining its basal level at 48 h p.t. (Figure 2)
Figure 2.

Compound 14 mediated up-regulation of Th1 cytokines (IL-12 and TNF-α), down-regulation of Th2 cytokines (IL-10 and TGF-β), and generation of nitric oxide in infected mouse macrophage (J-774A.1) cell line. Each bar represents pooled data of two individual experiments (mean ± SD value). Miltefosine (MF) is used as a reference drug. The asterisks indicate statistically significant increase and decrease (*, p < 0.05; **, p < 0.01) in cytokine production and NO generation compared between compound 14 treated versus untreated infected macrophages. ns, nonsignificant.
Enzyme assay along with physical studies is the best way to determine the mechanism of action of new compounds. However, high structural similarity between PTR1 inhibitors and present compounds has been considered for carrying out the physical investigation through hypothetical docking studies.27 The docking experiments were performed in Autodock 4.0.28 Prior to the docking, the molecule/ligand and protein structure were prepared by geometry optimization using a combination of the standard Tripos molecular mechanics force field of the SYBYL molecular modeling package29 with Powell energy minimization algorithm, Gasteiger–Huckel charges, and 0.001 kcal/(mol·Å) energy gradient convergence criterion. The Lamarckian genetic algorithm was used for docking the molecules. In docking experiments, pentamidine and the most potent molecule 14 occupied a location in the PTR1 binding pocket (PDB id: 1E7W), which is comparable to its ligand, methotrexate. Here, pentamidine (IC50 = 13.68 μM) and compound 14 (IC50 = 1.99 μM) showed −6.95 and −8.54 kcal/mol as binding energies, respectively. They shared Phe113, Tyr114, and Tyr191 as common binding residues. Figure 3 gives a 2D-view of the binding pocket interactions. The docking interactions indicated that compound 14 showed H-bonding and π-stacking with Tyr191 (Figure 4b). In the case of pentamidine, its interaction with Tyr191 is limited to π-stacking alone (Figure 4a). Along with compound 14, we also examined the docked poses of a less active compound (compound 17). Interestingly, in docking experiments, the less active compound did not mimic the active analogue (14) as well as reference compound pentamidine (Figure 4d). Moreover, compound 17 is protruding from the PTR1 binding domain (Figure 4c). These studies inferred that compound 14 is more potent compared to others due to its favorable hydrophobic and H-bond interactions with the binding domain of the PTR1.
Figure 3.
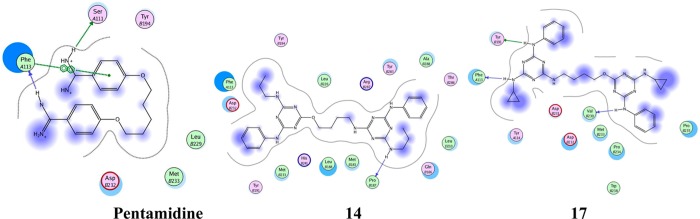
Two-dimensional view of pentamidine and compounds 14 and 17 with their common binding residues and possible hydrophobic interactions.
Figure 4.
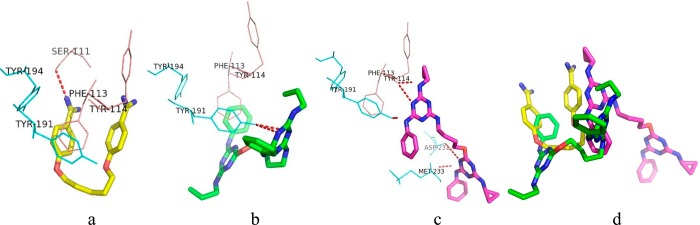
(a,b,c) Docked conformations of pentamidine and compounds 14 and 17. (d) Superimposition of pentamidine and the most active (14) and least active (17) compounds, which is used to differentiate the binding mode of the most active and least active compounds compared with the reference (pentamidine).
In conclusion, most of the synthesized compounds exhibited better potency against the intracellular amastigotes of L. donovani than the standard drug pentamidine and were not found to be cytotoxic. Compound 14 showed very consistent and promising leishmanicidal activity against intracellular amastigotes in vitro. This compound also displayed in vivo potential in the L. donovani/golden hamster model, as the hamster model of VL closely mimic the active human VL condition. Furthermore, compound 14 stimulates cell-mediated immune responses to prevent progression of leishmania parasite. Molecular docking analysis further validated the concepts involved in exploring these compounds. Interestingly, antileishmanial compounds also show activity against other protozoan parasites. It makes compound 14 more interesting for further investigation. These results give scope to further exploration of triazine mimetics as new antileishmanial leads.
Acknowledgments
We are thankful to S.A.I.F. Division of CDRI, Lucknow, for providing the spectroscopic data (CDRI Communication number: 8549).
Supporting Information Available
Final compounds characterization data; materials and methods of docking study associated with this article. This material is available free of charge via the Internet at http://pubs.acs.org.
We are grateful to Council of Scientific and Industrial Research (to K.C.) and Indian Council of Medical Research (to M.S.) for the financial support in the form of fellowships.
The authors declare no competing financial interest.
Supplementary Material
References
- Patil R. S.; Patil M. S.; Kshirsagar S. S.; Chaudhari P. S.; Bayas J. P.; Oswa R. J. Synthetic and natural products against leishmaniasis: A review. World J. Public Health Sci. 2012, 1, 7–22. [Google Scholar]
- Alvar J.; Canavate C.; Gutierrez-Solar B.; Jimenez M.; Lagnon F.; Lopez-Velet R.; Molina R.; Moreno J. Leishmania and human immunodeficiency virus coinfection: the first 10 years. Clin. Microbiol. Rev. 1997, 10, 298–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong I. L. K.; Chan K. F.; Chan T. H.; Chow L. M. C. Flavonoid dimers as novel, potent antileishmanial agents. J. Med. Chem. 2012, 55, 8891–8902. [DOI] [PubMed] [Google Scholar]
- Alexander J.; Bryson K. T helper (h)1/ Th2 and Leishmania: paradox rather than paradigm. Immunol. Lett. 2005, 99, 17–23. [DOI] [PubMed] [Google Scholar]
- Bogdon C.; Gessner A.; Solbach W.; Rollinghoff M. Invasion, control and persistence of Leishmania parasites. Curr. Opin. Immunol. 1996, 8, 517–525. [DOI] [PubMed] [Google Scholar]
- Stanley A. C.; Engwerda C. R. Balancing immunity and pathology in visceral leishmaniasis. Immunol. Cell Biol. 2007, 85, 138–147. [DOI] [PubMed] [Google Scholar]
- Diefenbach A.; Schiende H.; Rollinghoff M.; Yokoyama W. M.; Bogdan C. Requirement for type 2 NO synthase for Il–12 signaling in innate immunity. Science 1999, 284, 951–955. [DOI] [PubMed] [Google Scholar]
- Ashley J. N.; Barber H. J.; Ewins A. J.; Newbery G.; Self A. D. H. Chemotherapeutic comparison of the trypanocidal action of some aromatic diamidines. J. Chem. Soc. 1942, 103–116. [Google Scholar]
- Fairlamb A. H. Chemotherapy of human African trypanosomiasis: current and future prospects. Trends Parasitol. 2003, 19, 488–494. [DOI] [PubMed] [Google Scholar]
- Bouteille B.; Oukem O.; Bisser S.; Dumas M. Treatment perspectives for human African trypanosomiasis. Fundam. Clin. Pharmacol. 2003, 17, 171–181. [DOI] [PubMed] [Google Scholar]
- Croft S. L.; Yardley V. Chemotherapy of leishmaniasis. Curr. Pharm. Des. 2002, 8, 319–342. [DOI] [PubMed] [Google Scholar]
- Singh S.; Sivakumar R. Challenges and new discoveries in the treatment of leishmaniasis. J. Infect. Chemother. 2004, 10, 307–315. [DOI] [PubMed] [Google Scholar]
- Ismail M. A.; Arafa R. K.; Brun R.; Wilson W. D.; Generaux C.; Boykin D. W. Synthesis, DNA affinity, and antiprotozoal activity of linear dications: terphenyl diamidines and analogues. J. Med. Chem. 2006, 49, 5324–5332. [DOI] [PubMed] [Google Scholar]
- Bray P. G.; Barrett M. P.; Ward S. A.; de Koning H. P. Pentamidine uptake and resistance in pathogenic protozoa: past, present and future. Trends Parasitol. 2003, 19, 232–239. [DOI] [PubMed] [Google Scholar]
- Bakunov S. A.; Bakunova S. M.; Bridges A. S.; Wenzler T.; Barszcz T.; Werbovetz K. A.; Brun R.; Tidwell R. R. Synthesis and antiprotozoal properties of pentamidine congeners bearing the benzofuran motif. J. Med. Chem. 2009, 52, 5763–5767. [DOI] [PubMed] [Google Scholar]
- Zacharie B.; Abbott S. D.; Bienvenu J. F.; Cameron A. D.; Cloutier J.; Duceppe J.; Ezzitouni A.; Fortin D.; Houde K.; Lauzon C.; Moreau N.; Perron V.; Wilb N.; Asselin M.; Doucet A.; Fafard M. E.; Gaudreau D.; Grouix B.; Bournet F. S.; St-Amant N.; Gagnon L.; Penney C. L. 2,4,6-Trisubstituted triazines as protein A mimetics for the treatment of autoimmune diseases. J. Med. Chem. 2010, 53, 1138–1145. [DOI] [PubMed] [Google Scholar]
- Porwal S.; Chauhan S. S.; Chauhan P. M. S.; Shakya N.; Verma A.; Gupta S. Discovery of novel antileishmanial agents in an attempt to synthesize pentamidine–aplysinopsin hybrid molecule. J. Med. Chem. 2009, 19, 5793–5802. [DOI] [PubMed] [Google Scholar]
- Tyagi V.; Khan S.; Shivahare R.; Srivastava K.; Gupta S.; Kidwai S.; Srivastava K.; Puri S. K.; Chauhan P. M. S. A natural product inspired hybrid approach towards the synthesis of novel pentamidine based scaffolds as potential anti-parasitic agents. Bioorg. Med. Chem. Lett. 2013, 23, 291–296. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Chauhan K.; Shivahare R.; Vishwakarma P.; Suthar M. K.; Sharma A.; Gupta S.; Saxena J. K.; Lal J.; Chandra P.; Kumar B.; Chauhan P. M. S. Discovery of a new class of natural product-inspired quinazolinone hybrid as potent antileishmanial agents. J. Med. Chem. 2013, 56, 4374–4392. [DOI] [PubMed] [Google Scholar]
- Gangjee A.; Jain H. D.; Kurup S. Recent advances in classical and nonclassical antifolates as antitumor and antiopportunistic infection agents: Part II. Anticancer Agents Med. Chem. 2008, 8, 205–231. [DOI] [PubMed] [Google Scholar]
- Cunningham M. L.; Beverley S. M. Pteridine salvage throughout the Leishmania infectious cycle: implication for antifolate chemotherapy. Mol. Biochem. Parasitol. 2001, 113, 199–213. [DOI] [PubMed] [Google Scholar]
- McLuskey K.; Gibellini F.; Carvalho P.; Avery M. A.; Hunter W. N. Inhibition of Leishmania major pteridine reductase by 2,4,6-triaminoquinazoline: structure of the NADPH ternary complex. Acta Crystallogr. 2004, 60, 1780–1785. [DOI] [PubMed] [Google Scholar]
- Corona P.; Gibellini F.; Cavalli A.; Saxena P.; Carta A.; Loriga M.; Luciani R.; Paglietti G.; Guerrieri D.; Nerini E.; Gupta S.; Hannaert V.; Michels P. A. M.; Ferrari S.; Costi P. M. Structure-based selectivity optimization of piperidine–pteridine derivatives as potent Leishmania pteridine reductase inhibitors. J. Med. Chem. 2012, 55, 8318–8329. [DOI] [PubMed] [Google Scholar]
- Kaur J.; Sundar S.; Singh N. Molecular docking, structure–activity relationship and biological evaluation of the anticancer drug monastrol as a pteridine reductase inhibitors in a clinical isolates of Leishmania donovani. J. Antimicrob. Chemother. 2010, 65, 1742–1748. [DOI] [PubMed] [Google Scholar]
- Chauhan K.; Sharma M.; Saxena J.; Singh S. V.; Trivedi P.; Srivastava K.; Puri S. K.; Saxena J. K.; Chaturvedi V.; Chauhan P. M. S. Synthesis and biological evaluation of a new class of 4-aminoquinoline–rhodanine hybrid as potent anti-infective agents. Eur. J. Med. Chem. 2013, 62, 693–704. [DOI] [PubMed] [Google Scholar]
- Chauhan K.; Sharma M.; Singh P.; Kumar V.; Shukla P. K.; Siddiqi M. I.; Chauhan P. M. S. Discovery of a new class of dithiocarbamates and rhodanine scaffold as potent antifungal agents: Synthesis, biology and molecular docking. Med. Chem. Commun. 2012, 3, 1104–1110. [Google Scholar]
- Mpamhanga C. P.; Spinks D.; Tulloch L. B.; Shanks E. J.; Robinson D. A.; Collie I. T.; Fairlamb A. H.; Wyatt P. G.; Frearson J. A.; Hunter W. N.; Gilbert I. H.; Brenk R. One scaffold, three binding modes: Novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J. Med. Chem. 2009, 52, 4454–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SYBYL, version 7.3; Tripos Associates: St. Louis MO, 2006.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.