

Abstract
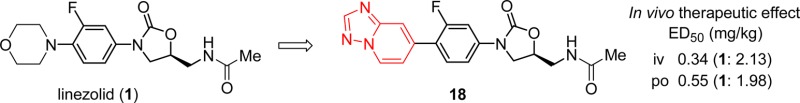
Novel oxazolidinone analogues bearing a condensed heteroaromatic ring as the C-ring substructure were synthesized as candidate antibacterial agents. Analogues 16 and 21 bearing imidazo[1,2-a]pyridine and 18 and 23 bearing [1,2,4]triazolo[1,5-a]pyridine as the C-ring had excellent in vitro antibacterial activities against methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus faecalis (VRE), and penicillin-resistant Streptococcus pneumoniae (PRSP). They also showed promising therapeutic effects in a mouse model of lethal infection. Preliminary safety data (inhibitory effects on cytochrome P450 isoforms and monoamine oxidases) were satisfactory. Further evaluation of 18 and 23 is ongoing.
Keywords: Antibacterial, oxazolidinone, methicillin-resistant Staphylococcus aureus, condensed heteroaromatic ring
Infections caused by multidrug-resistant organisms, such as methicillin-resistant Staphylococcus aureus (MRSA),1 vancomycin-resistant Enterococcus faecalis (VRE),2 and penicillin-resistant Streptococcus pneumoniae (PRSP),3 are a major problem in hospitals around the world, and effective antibiotics are urgently needed. Oxazolidinone antibacterials are a class of synthetic antibiotics that have been developed over the last three decades4,5 and are composed of an A-ring (oxazolidinone), B-ring (phenyl), and C-ring (an appropriate heterocycle, such as morpholine or piperazine), with a C-5 side chain unit on the A-ring substructure. Linezolid (1) was the first oxazolidinone antibiotic to be approved by the US Food and Drug Administration (FDA) and has been used for the treatment of serious infections caused by Gram-positive strains such as MRSA and VRE since 2000 (Figure 1).6 However, linezolid-resistant Staphylococcus aureus and Enterococcus spp.7−9 have appeared since 1 was released, and 1 is also an inhibitor of monoamine oxidases (MAOs), which may result in drug–drug interactions with adrenergic and serotonergic agents.10 Thus, there is considerable interest in new oxazolidinone-type drug candidates (Figure 1). A series of oxazolidinones containing a pyrroloaryl substituent as C-ring unit was developed by J&J Pharmacuetical, and one analogue advanced to phase I clinical trial.11 Ranbezolid (2), which has a furan ring as the terminal unit, was developed by Ranbaxy Laboratories, and a phase I clinical trial was performed in 2003.12,13 Radezolid (3), which has a [1,2,3]triazole framework as the terminal substructure, was developed by Rib-X Pharmaceuticals and has completed a phase II clinical trial.5 Tedizolid phosphate (4, formerly called torezolid phosphate), which contains a tetrazole structure, was developed by Trius Therapeutics and has recently completed a phase III clinical trial.14 These candidate compounds all have a heteroaromatic ring as the terminal chemical structure unit. However, Okamoto et al. reported the synthesis and in vitro antimycobacterial activity of pyrazolo[1,5-a]pyridine derivatives and suggested that the condensed heteroaromatic ring system was a pharmacophore exhibiting antimycobacterial activity.15 Although this scaffold has not been investigated for activity against Gram-positive or -negative bacteria, the oxazolidinone antibacterial sutezolid (5) is known to exhibit strong in vitro activities against both Gram-positive bacteria (such as MRSA) and Mycobacterium tuberculosis.16 Since the above heteroaromatic ring system showed antimycobacterial activity, we suspected that it might also have antibacterial activity potential because antimycobacterial activity is generally correlated with antibacterial activity toward Gram-positive bacteria such as MRSA. Therefore, we expected that introduction of the above pyrazolo[1,5-a]pyridine or a related scaffold into oxazolidinone antibacterials might result in potent antibacterial activity. Herein, we report the synthesis and biological evaluations of novel oxazolidinone derivatives bearing pyrazolo[1,5-a]pyridine or its similar scaffold as the C-ring substructure.
Figure 1.

Recently developed potent antibacterials of the oxazolidinone family.
The synthesis of the desired oxazolidinone analogues was performed by Suzuki–Miyaura coupling reaction of the known boronic acid ester (6)17 with 6-bromopyrazolo[1,5-a]pyridine (7),18 6-bromoimidazo[1,5-a]pyridine (8),19 6-bromoimidazo[1,2-a]pyridine (9),20 7-bromoimidazo[1,2-a]pyridine (10),21 6-bromo[1,2,4]triazolo[1,5-a]pyridine (11),22 and 7-bromo[1,2,4]triazolo[1,5-a]pyridine (12),23 as shown in Table 1. The products 13–18 were recrystallized from appropriate organic solvents. These compounds 13–18 were then evaluated for in vitro antibacterial activity against Gram-positive (S. aureus, E. faecalis, E. faecium, and S. pneumoniae) and Gram-negative (Moraxella catarrhalis and Haemophilus influenzae) bacteria using a conventional broth microdilution method. Activity against linezolid-resistant S. aureus NRS271, a clinical isolate, was also examined. NRS271 carries G2576T mutation in domain V, the peptidyl transferase center, of 23S rRNA gene (Escherichia coli 23S rRNA numbering). As shown in Table 2, the minimum inhibitory concentrations (MICs, μg/mL) of 13–18 against S. aureus generally indicated 2- to 8-fold greater potency in comparison with linezolid 1. Compounds 13 and 16 showed clinically relevant MIC values against linezolid-resistant S. aureus NRS271, as well as Gram-negative M. catarrhalis. Compound 16 with an imidazo[1,2-a]pyridine ring unit also exhibited good antibacterial activity toward Gram-negative H. influenzae. Next, we examined the effect of replacing the conventional acetamide type C-5 side chain unit on the A-ring with a [1,2,3]triazole unit, which has been reported to enhance antibacterial activity and reduce inhibition of monoamine oxidase (MAO).24 The compounds were synthesized via a similar procedure to that described for compounds 13–18, using boronic acid ester (19)25 instead of 6, with heteroaryl bromides 9–12 (Table 3). The prepared compounds 20–23 exhibited similar in vitro antibacterial activity to the acetamide derivatives 15–18, and compound 21 showed most potent antibacterial activity among all the synthesized compounds (Table 4). We then evaluated the in vivo therapeutic effect of selected compounds via intravenous or oral administration in a mouse lethal systemic infection model employing S. aureus SR3637 (MRSA). The results are shown in Table 5. Though compound 13 had potent in vitro antibacterial activity, it was not effective in vivo. This unsuccessful result may suggest that 13 has poor pharmacokinetic (PK) properties or is metabolically unstable. However, other tested compounds showed moderate to excellent in vivo therapeutic effect, regardless of administration route. Compound 18 exhibited a 4- to 6-fold greater in vivo potency than that of linezolid 1 via both intravenous and oral administration, in line with the in vitro results. In addition, compounds 16 and 21, bearing an imidazo[1,2-a]pyridine C-ring unit, also showed a good therapeutic effect, superior to that of linezolid, after both intravenous and oral administration. Compound 23 also had a good therapeutic effect via oral administration. The pharmacokinetics (PK) of compound 18 was evaluated after oral administration to mice (Table 6). Compound 18 showed a sufficient AUC even at a 10-fold lower dosage than linezolid (1), used as a reference compound. This excellent PK profile is consistent with the in vivo efficacy of 18 regardless of the route of administration. We made a preliminary examination of the safety profile of the in vivo active compounds 16, 18, 21, and 23. Specifically, we examined whether these compounds inhibit four subtypes of cytochrome P450 (CYP) isoforms, as well as MAO-A, and MAO-B. The results are listed in Table 7. Three compounds did not show marked CYP inhibition even at concentrations greatly exceeding the antibacterial minimum inhibitory concentrations, and their inhibition of MAO-A was less than that of linezolid 1, though compound 18 inhibited MAO-B slightly more strongly than 1. Compound 21 with the [1,2,3]triazole C-5 side chain unit on the oxazolidinone ring showed low inhibition rates of MAO-A and MAO-B. Furthermore, compound 23 exhibited the lowest inhibition rate toward MAO-A among the four tested compounds. On the basis of these results, we consider that compounds 18 and 23 are promising candidates for more extensive evaluation. In particular, it will be important to evaluate the toxicity of these compounds, particularly myleosuppression and peripheral neuropathy, because this is often an issue with oxazolidinone-type antibiotics. In the present work, we introduced six kinds of basic condensed heteroaromatic ring substructure as the C-ring part and obtained potent compounds 18 and 23, with promising in vitro and in vivo antibacterial efficacy. We are now examining in more detail the structure–activity relationships around compounds 18 and 23, which represent both lead compounds and potential clinical candidates.
Table 1. Synthesis of Targeted Oxazolidinone Analogues 13–18 Bearing an Acetamide C-5 Side Chain on the Oxazolidinone Ring.
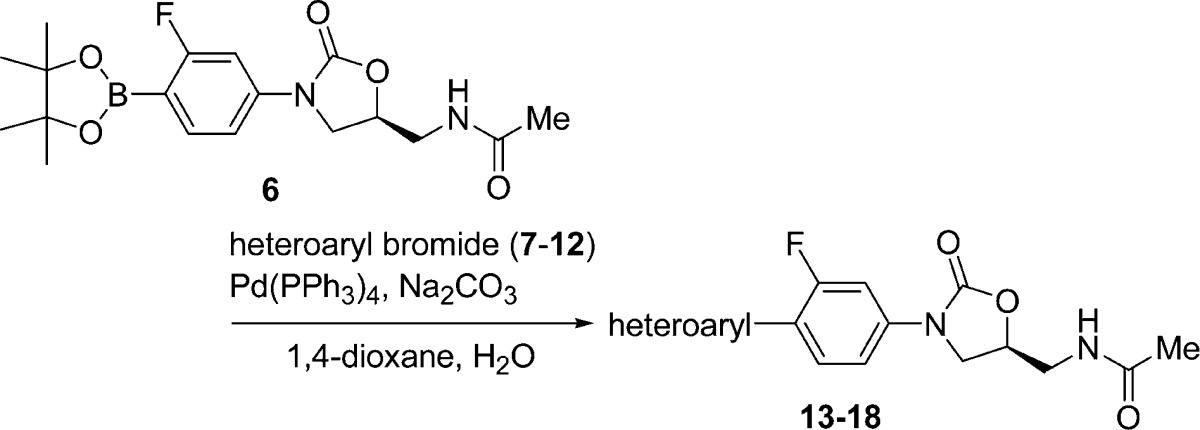
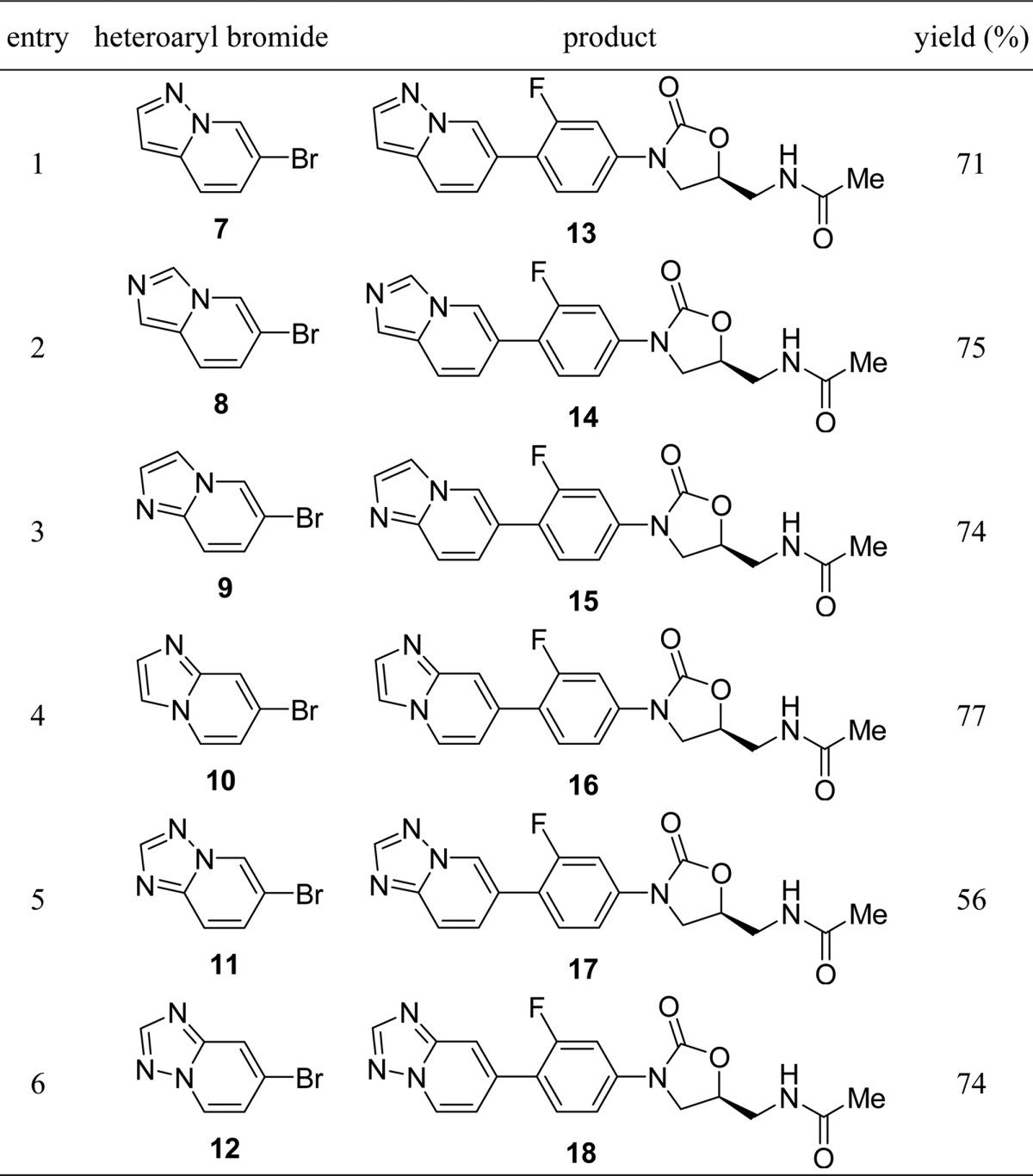
Table 2. In Vitro Antibacterial Activity of Compounds 13–18 Bearing an Acetamide C-5 Side Chain on the Oxazolidinone Ring.
| minimum inhibitory concentration (μg/mL) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd | S.a.a | S.a.b | S.a.c | S.a.d | E.f.e | E.f.f | M.c.g | S.p.h | S.p.i | H.i.j |
| 1 | 2 | 2 | 2 | 32 | 4 | 2 | 8 | 1 | 0.5 | 16 |
| 13 | 0.25 | 0.5 | 0.25 | 2 | 0.5 | 0.25 | 2 | ≤0.063 | ≤0.063 | 8 |
| 14 | 0.25 | 1 | 0.5 | 4 | 0.5 | 0.5 | 4 | ≤0.063 | ≤0.063 | 8 |
| 15 | 0.5 | 1 | 0.5 | 8 | 0.5 | 0.25 | 8 | ≤0.063 | ≤0.063 | 4 |
| 16 | 0.25 | 0.25 | 0.25 | 2 | 0.25 | 0.5 | 2 | ≤0.063 | ≤0.063 | 2 |
| 17 | 0.5 | 0.5 | 0.5 | 8 | 0.5 | 0.5 | 8 | ≤0.063 | ≤0.063 | 4 |
| 18 | 0.25 | 0.5 | 0.25 | 4 | 0.25 | 0.25 | 4 | ≤0.063 | ≤0.063 | 4 |
Staphylococcus aureus SR20549.
Staphylococcus aureus Smith.
Staphylococcus aureus SR3637 (methicillin-resistant).
Staphylococcus aureus NRS271(linezolid-resistant).
Enterococcus faecalis SR1004.
Enterococcus faecium SR7940 (vancomycin-resistant).
Moraxella catarrhalis SR26840.
Streptococcus pneumonoiae SR26207.
Streptococcus pneumonoiae SR11031 (penicillin-resistant).
Haemophilus influenzae SR27914.
Table 3. Synthesis of Targeted Oxazolidinone Analogues 20–23 Bearing a [1,2,3]Triazole C-5 Side Chain on the Oxazolidinone Ring.
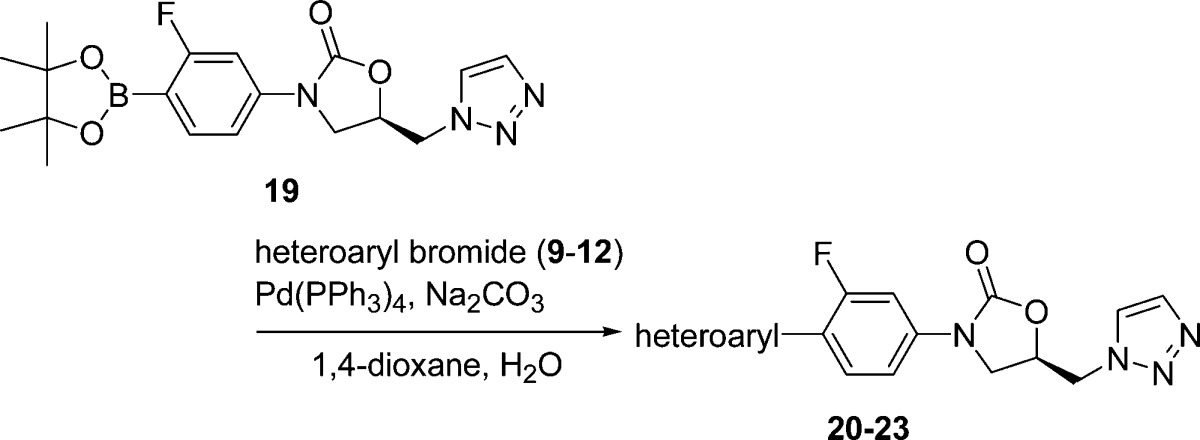
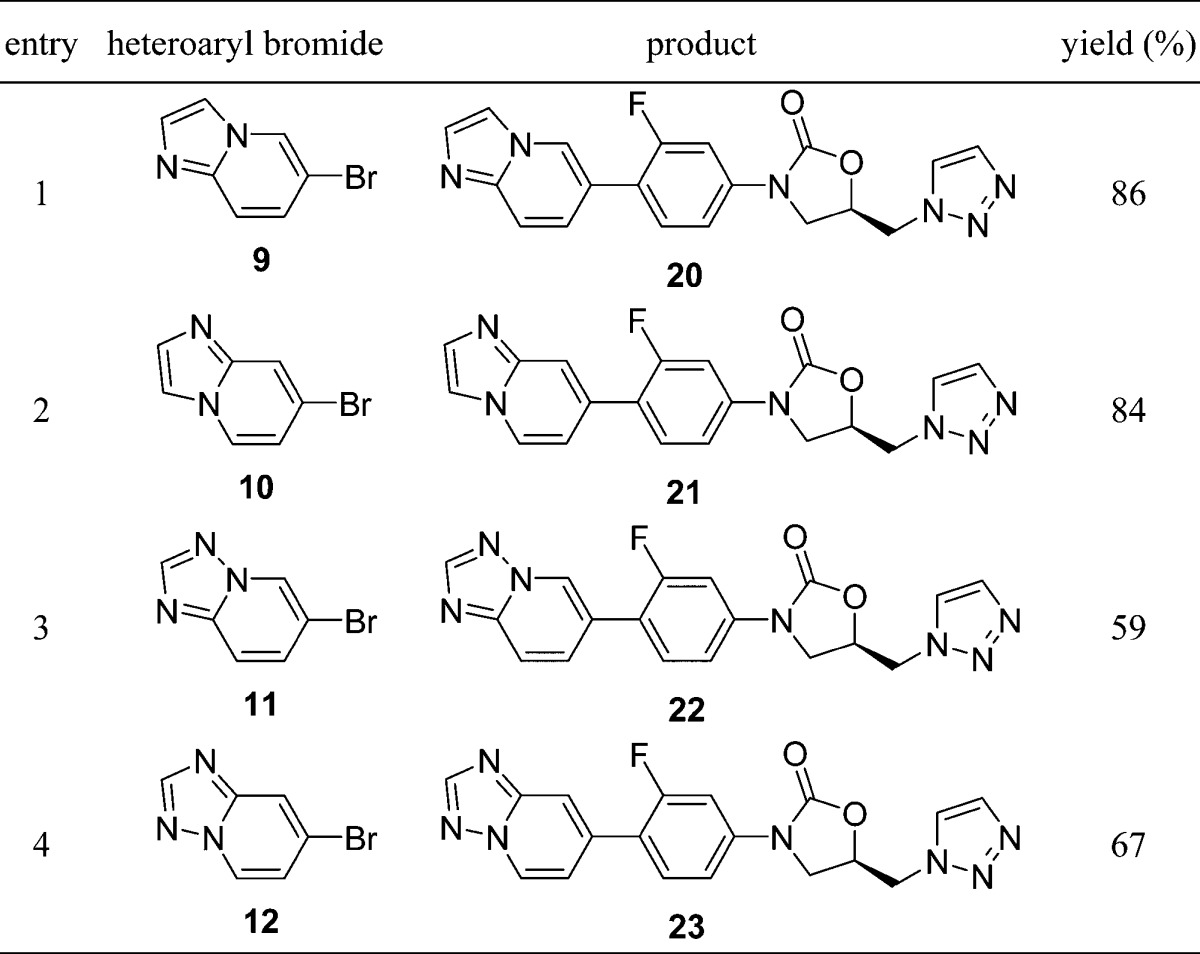
Table 4. In Vitro Antibacterial Activity of Compounds 20–23 Bearing a [1,2,3]Triazole C-5 Side Chain on the Oxazolidinone Ring.
| minimum inhibitory concentration (μg/mL) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd | S.a.a | S.a.b | S.a.c | S.a.d | E.f.e | E.f.f | M.c.g | S.p.h | S.p.i | H.i.j |
| 1 | 2 | 2 | 2 | 32 | 4 | 2 | 8 | 1 | 0.5 | 16 |
| 20 | 0.25 | 0.5 | 0.25 | 4 | 0.5 | 0. 5 | 4 | ≤0.063 | ≤0.063 | 4 |
| 21 | 0.125 | 0.25 | 0.125 | 2 | 0.25 | 0.25 | 2 | ≤0.063 | ≤0.063 | 2 |
| 22 | 0.25 | 0.5 | 0.25 | 8 | 0.5 | 0.25 | 8 | ≤0.063 | ≤0.063 | 4 |
| 23 | 0.125 | 0.25 | 0.25 | 2 | 0.25 | 0.25 | 4 | ≤0.063 | ≤0.063 | 2 |
Staphylococcus aureus SR20549.
Staphylococcus aureus Smith.
Staphylococcus aureus SR3637 (methicillin-resistant).
Staphylococcus aureus NRS271(linezolid-resistant).
Enterococcus faecalis SR1004.
Enterococcus faecium SR7940 (vancomycin-resistant).
Moraxella catarrhalis SR26840.
Streptococcus pneumonoiae SR26207.
Streptococcus pneumonoiae SR11031 (penicillin-resistant).
Haemophilus influenzae SR27914.
Table 5. In Vivo Therapeutic Effects of Selected Oxazolidinone Analogues in a Mouse Lethal Systemic Infection Model Employing S. aureus SR3637.
| therapeutic effect ED50 (mg/kg)a |
|||
|---|---|---|---|
| administration route |
|||
| compd | MIC (μg/mL) | iv | po |
| 13 | 0.25 | >10 (1: 1.98) | N.T. |
| 14 | 0.5 | 3.05 (1: 4.32) | N.T. |
| 15 | 0.5 | 3.17 (1: 2.46) | 3.05 (1: 2.83) |
| 16 | 0.25 | 1.09 (1: 1.73) | 1.05 (1: 2.83) |
| 18 | 0.25 | 0.34 (1: 2.13) | 0.55 (1: 1.98) |
| 21 | 0.125 | 0.95 (1: 1.73) | 1.02 (1: 2.83) |
| 23 | 0.25 | N.T. | 0.95 (1: 3.87) |
Compound 1 was used as reference in each test, and its ED50 value was shown in the parentheses. N.T.: not tested.
Table 6. PK Properties of in Vivo Potent Analogue 18 in Mice.
| parameter | linezolid (1) | 18 |
|---|---|---|
| dose (mg/kg) | 67.7 | 5.56 |
| Tmax (hr) | 0.50 | 1.00 |
| Cmax (μg/mL) | 37.00 | 4.78 |
| AUC∞ (μg·h/mL) | 161.85 | 30.93 |
Table 7. CYP450 Isoform and MAO-A,B Inhibition Indices of in Vivo-Active Analogues 16, 18, 21, and 23.
| CYP450 isoform, IC50 (μM) |
MAO inhibition (%) (30 μM
of each compound) |
|||||
|---|---|---|---|---|---|---|
| compd | 1A2 | 2C9 | 2D6 | 3A4 | A | B |
| 1 | >20 | >20 | >20 | >20 | 61.3 | 70.4 |
| 16 | >20 | >20 | >20 | >20 | 54.8 | 52.8 |
| 18 | >20 | >20 | >20 | >20 | 55.9 | 78.2 |
| 21 | >20 | >20 | >20 | 15 | 41.2 | 26.7 |
| 23 | >20 | >20 | >20 | >20 | 33.2 | 27.9 |
In summary, we have synthesized several oxazolidinone-type antibacterials bearing a condensed heteroaromatic ring system as the C-ring and evaluated their activity against a panel of multidrug-resistant organisms. Almost all of the synthesized compounds exhibited potent in vitro antibacterial activity and several also showed superior in vivo antibacterial activity in a mouse model of lethal infection. The in vivo-active compounds 16, 18, 21, and 23 also showed similar or improved values of MAO-A inhibition index in comparison with linezolid 1, along with similar levels of CYP inhibition potential (IC50 >10 μM for the four selected isoforms) to 1, and these levels are satisfactory from a safety point of view. More detailed evaluation of the potent compounds 18 and 23 is ongoing, together with synthesis of further analogues for structure–activity relationship studies.
Acknowledgments
We are grateful to Professor Hiroyuki Kagechika and Dr. Hiroyuki Masuno of the Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University, for measurement of EI-LRMS and HRMS spectra. Linezolid-resistant strain NRS271 was kindly provided by the network on antimicrobial resistance in Staphylococcus aureus (http://www.narsa.net/content/default.jsp).
Supporting Information Available
Synthetic method, characterization of compounds, and protocols of biological evaluations. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This paper posted ASAP on September 24, 2013. A correction was made to Table 7 and the revised version was reposted on September 25, 2013.
Supplementary Material
References
- Ender M.; McCallum N.; Adhikari R.; Berger-Bächi B. Fitness cost of SCCmec and methicillin resistance levels in Staphylococcus aureus. Antimicrob. Agents Chemother. 2004, 48, 2295–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttley A. H. C.; Collins C. H.; Naidoo J.; George R. C. Vancomycin-resistant enterococci. Lancet 1988, 1, 57–58. [DOI] [PubMed] [Google Scholar]
- Goldstein F. W. Penicillin-resistant Streptococcus pneumoniae: selection by both β-lactam and non-β-lactam antibiotics. J. Antimicrob. Chemother. 1999, 44, 141–144. [DOI] [PubMed] [Google Scholar]
- Brickner S. J. Oxazolidinone antibacterial agents. Curr. Pharm. Des. 1996, 2, 175–194. [Google Scholar]
- Michalska K.; Karpiuk I.; Krol M.; Tyski S. Recent development of potent analogues of oxazolidinone antibacterial agents. Bioorg. Med. Chem. 2013, 21, 577–591. [DOI] [PubMed] [Google Scholar]
- Moellering R. C. Jr. Linezolid: The first oxazolidinone antimicrobial. Drugs Drug Ther. 2003, 138, 135–142. [DOI] [PubMed] [Google Scholar]
- Tsiodras S.; Gold H. S.; Sakoulas G.; Eliopoulos G. M.; Wennersten C.; Venkataraman L.; Moellering R. C.; Ferraro M. J. Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet 2001, 358, 207–208. [DOI] [PubMed] [Google Scholar]
- Gonzales R. D.; Schreckenberger P. C.; Graham M. B.; Kelkar S.; Den-Besten K.; Quinn J. P. Infections due to vancomycin-resistant Enterococcus faecium resistant to linezolid. Lancet 2001, 357, 1179. [DOI] [PubMed] [Google Scholar]
- Toh S.; Xiong L.; Arias C. A.; Villegas V.; Lolans K.; Quinn J.; Mankin A. S. Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol. Microbiol. 2007, 64, 1506–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renslo A. R. Antibacterial oxazolidinones: emerging structure–toxicity relationships. Expert Rev. Anti-Infect. Ther. 2010, 8, 565–574. [DOI] [PubMed] [Google Scholar]
- Paget S. D.; Foleno B. D.; Boggs C. M.; Goldschmidt R. M.; Hlasta D. J.; Weidner-Wells M. A.; Werblood H. M.; Wira E.; Bush K.; Macielag M. J. Synthesis and antibacterial activity of pyrroloaryl-substituted oxazolidinones. Bioorg. Med. Chem. Lett. 2003, 13, 4173–4178. [DOI] [PubMed] [Google Scholar]
- Das B.; Rundra S.; Yadav A.; Ray A.; Raja Rao A. V. S.; Srinivas A. S. S. V.; Soni A.; Saini S.; Shukla S.; Pandya M.; Bhateja P.; Malhotra S.; Mathur T.; Arora S. K.; Rattan A.; Mehta A. Synthesis and SAR of novel oxazolidinones: Discovery of ranbezolid. Bioorg. Med. Chem. Lett. 2005, 15, 4261–4267. [DOI] [PubMed] [Google Scholar]
- Bambeke F. V.; Reinert R. R.; Appelbaum P. C.; Tulkens P. M.; Peetermans W. E. Multidrug-resistant Streptococcus pneumoniae infections. Drugs 2007, 67, 2355–2382. [DOI] [PubMed] [Google Scholar]
- Philippe P.; Carisa D. A.; Edward F.; Purvi M.; Anita D. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections. The ESTABLISH-1 randomized trial. J. Am. Med. Assoc. 2013, 309, 559–569. [DOI] [PubMed] [Google Scholar]
- Suzue S.; Hirobe M.; Okamoto T. Synthetic antimicrobials. II. Synthesis of pyrazolo[1,5-a]pyridine derivatives. (1). Chem. Pharm. Bull. 1973, 21, 2146–2160. [Google Scholar]
- Villemagne B.; Crauste C.; Flipo M.; Baulard A. R.; Deprez B.; Willand N. Tuberculosis: The drug development pipeline at a glance. Eur. J. Med. Chem. 2012, 51, 1–16. [DOI] [PubMed] [Google Scholar]
- Hales N. J.; Weber T. P.; Gravestock M. B.; Carcanague D. R.; Hauck S. I.. Oxazolidinone and/or isoxazoline derivatives as antibacterial agents. PCT Int. Appl. WO200448392, 2004.
- Kendall J. D.; Giddens A. C.; Tsang K. Y.; Frédérick R.; Marshall E. S.; Singh R.; Lill C. L.; Lee W.-J.; Kolekar S.; Chao M.; Malik A.; Yu S.; Chaussade C.; Buchanan C.; Rewcastle G. W.; Baguley B. C.; Flanagan J. U.; Jamieson S. M. F.; Denny W. A.; Shepherd P. R. Novel pyrazolo[1,5-a]pyridines as p110α-selective PI3 kinase inhibitors: Exploring the benzenesulfonohydrazide SAR. Bioorg. Med. Chem. 2012, 20, 58–68. [DOI] [PubMed] [Google Scholar]
- Chytil M.; Engel S. R.; Fang Q. K.; Spear K. L.. Histamine H3 inverse agonists and antagonists and methods of use thereof. PCT Int. Appl. WO201131818, 2011.
- Yamanaka M.; Miyake K.; Suda S.; Ohhara H.; Ogawa T. Imidazo[1,2-a]pyridines. I. Synthesis and inotropic activity of new 5-imidazo[1,2-a]pyridinyl-2(1H)-pyridinone derivatives. Chem. Pharm. Bull. 1991, 39, 1556–1567. [DOI] [PubMed] [Google Scholar]
- Allen S.; Schlachter S. T.; Lyssikatos J. P.; Zhao Q.; Topalov G. T.; Robinson J. E.; Greschuk J. M.; Marmsaeter F. P.; Munson M. C.; Rizzi J. P.; Kallan N. C. Imidazo[1,2-A]pyridine compounds as receptor tyrosine kinase inhibitors. PCT Int. Appl. WO2008124323, 2008.
- Edmondson S. D.; Mastracchio A.; Mathvink R. J.; He J.; Harper B.; Park Y.-J.; Beconi M.; Salvo J. D.; Eiermann G. J.; He H.; Leiting B.; Leone J. F.; Levorse D. A.; Lyons K.; Patel R. A.; Patel S. B.; Petrov A.; Scapin G.; Shang J.; Roy R. S.; Smith A.; Wu J. K.; Xu S.; Zhu B.; Thornberry N. A.; Weber A. E. (2S,3S)-3-Amino-4-(3,3-difluoropyrrolidin-1-yl)-N,N-dimethyl-4-oxo-2-(4-[1,2,4]triazolo[1,5-a]- pyridin-6-ylphenyl)butanamide: A selective α-amino amide dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2006, 49, 3614–3627. [DOI] [PubMed] [Google Scholar]
- Baerfacker L.; Kast R.; Griebenow N.; Meier H.; Kolkhof P.; Albrecht-Kuepper B.; Nitsche A.; Stasch J.-P.; Schneider D.; Teusch N.; Rudolph J.; Whelan J.; Bullock W.; Pleasic-Williams S.. Substituted 5-aminopyrazoles and use thereof. PCT Int. Appl. WO201020363, 2010.
- Reck F.; Zhou F.; Girardot M.; Kern G.; Eyermann C. J.; Hales N. J.; Ramsay R. R.; Gravestock M. B. Identification of 4-substituted 1,2,3-triazoles as novel oxazolidinone antibacterial agents with reduced activity against monoamine oxidase A. J. Med. Chem. 2005, 48, 499–506. [DOI] [PubMed] [Google Scholar]
- Carcanague D. R.; Gravestock M. B.. 3-[4-(6-{4,5-Dihydroisoxazol-3-yl}pyridin-3-yl)-3-phenyl]-5-(1H- 1,2,3-triazol-1-ylmethyl)-1,3-oxazolidin-2-ones as antibacterial sgents. PCT Int. Appl. WO2005116021, 2005.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.