

Abstract
In this letter, we describe the design, synthesis, and structure–activity relationship of 5-anilinopyrazolo[1,5-a]pyrimidine inhibitors of CK2 kinase. Property-based optimization of early leads using the 7-oxetan-3-yl amino group led to a series of matched molecular pairs with lower lipophilicity, decreased affinity for human plasma proteins, and reduced binding to the hERG ion channel. Agents in this study were shown to modulate pAKTS129, a direct substrate of CK2, in vitro and in vivo, and exhibited tumor growth inhibition when administered orally in a murine DLD-1 xenograft.
Keywords: CK2 kinase; pyrazolo[1,5-a]pyrimidine; matched molecular pair; oxetane
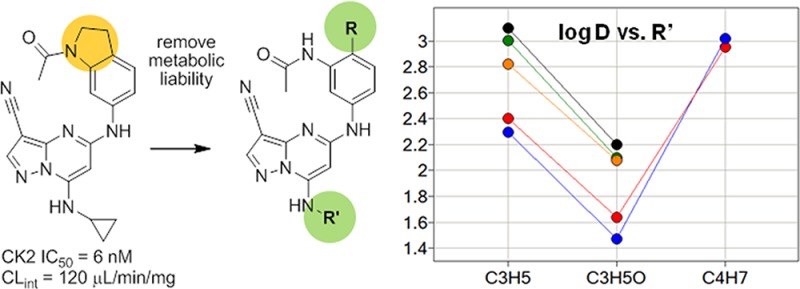
The serine/threonine protein kinase CK2, a tetrameric complex containing two catalytic (α or α′) and two regulatory (β) subunits, controls cell growth, proliferation, and evasion of apoptosis by phosphorylation of a range of substrates in critical cellular signaling pathways including PI3K (phosphatidylinositol 3-kinase)/AKT (protein kinase B), NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells) and Wnt (wingless type MMTV integration site family).1−3 Overexpression of the CK2α subunit correlates with tumor aggressiveness and disease severity in certain cancers, while compensatory increases in CK2α′ levels have been observed in response to RNAi treatment in mice.4,5 Several academic and industrial research groups have been actively engaged in developing small molecule inhibitors of CK2 to provide further pharmacological validation of the compelling in vitro and in vivo data amassed to date.6 The recent discovery of CX-4945, a selective, orally available inhibitor of CK2 by researchers from Cylene, represents an important first step in evaluating the clinical potential of this novel target in man.7 We have recently described the design of a series of conformationally constrained inhibitors of CK2 containing the pyrazolo[1,5-a]pyrimidine nucleus.8 Members of this series of compounds exhibited potent inhibition of the enzyme, possessed a high degree of kinase selectivity, and depleted cellular levels of pAKTS129, a direct substrate of CK2 believed to hyperactivate the AKT pathway.9 Although our attempts to enhance both cellular potency and physical properties in this series were unsuccessful, these studies resulted in an understanding of the structure–property relationships within the scaffold and provided additional insights into ligand–receptor binding. In particular, we found that N-methylation of the acetamide of 1a, to give ring-constrained analogue 1b, preserved enzymatic and cellular activity. In subsequent designs, we proposed to release the constraint in 1b and introduce a new conformational constraint, exemplified by indoline 2, that would enforce the crystallographically observed cisoid conformation of the acetamide in 1 (Figure 1). Compound 2 is a potent inhibitor of CK2 in biochemical and cellular assays; however, it exhibits low aqueous solubility at physiological pH (sol. = 4 μM) and high intrinsic clearance when incubated with rat liver microsomes (CLint = 120 μL/min/mg). We postulated that either the N-acetyl group or the methylene groups within the newly introduced five-membered ring represented potential sites of metabolism and chose instead to focus on other variations of the pharmacophore. The potency of 2 established that the position ortho to the acetamide could be functionalized, and we subsequently prepared a series of N-(3-aminoaryl)acetamide analogues (3) to evaluate the scope of substituents that might be accommodated on the aryl ring (Figure 1). Herein, we describe our efforts to build on the enzymatic potency and kinase selectivity profile of 1a, while striving for an acceptable balance of physical properties and ADME characteristics that would enable us to carry out pharmacodynamic and disease model studies using an orally administered agent.
Figure 1.

Design of conformationally constrained acetylindoline 2 and ortho-substituted anilines (3).
The synthesis of 2 and related analogues was carried out by adapting the convergent approach described in our earlier work (Scheme 1).8 Installation of functionality at C7 was accomplished by displacement of the 7-chloro group in 5,7-dichloro-3-cyano-pyrazolo[1,5-a]pyrimidine (4) to afford intermediates of general structure 5. Substitution at C5 was subsequently achieved by palladium catalyzed10 or KF-promoted coupling of 5 with the requisite N-(3-aminoaryl)acetamide (6) to furnish the desired analogues (7a–l).
Scheme 1.

Introduction of N-(3-aminophenyl)acetamides bearing chloro (7a) and small alkyl groups (7b–d) at the 6-position increased the half-maximum inhibitory potency below the limit of detection (IC50 < 3 nM) in our enzymatic assay, which measures the inhibition of recombinant human full length CK2α mediated phosphorylation of a synthetic peptide substrate at Km ATP concentration (Table 1).11 Among the C6 alkyl substituted derivatives, the methyl (7b) isopropyl (7c) and cyclopropyl (7d) analogues exhibit comparable activity in a mechanistic cellular assay that measures levels of the CK2 substrate pAKTS129 in an ELISA format. Introduction of a nitrogen atom in the aniline ring, to afford pyridyl analogues 7g and 7h, reduced cellular potency relative to 7b, with a negligible effect on solubility. We subsequently evaluated the effect of varying the substituent at the hinge-binding N7 position and found that five-membered heterocyclic substituents (7k,l) preserved enzymatic activity but, with the exception of N-methyl imidazole (7l), adversely affected solubility. With the enzymatic and mechanistic cellular activity of compounds in this series routinely reading out at or below the lower limits of detection in the respective assays, we began to guide our structure–activity relationship (SAR) studies with the aid of phenotypic assays using colorectal (HCT-116, DLD-1) and other cultured cell lines.
Table 1. CK2 Biochemical and Cellular Potency for Compounds 7a–l.
| compd | CK2α IC50 (nM)a | pAKTS129 IC50 (nM)a | HCT-116 GI50 (nM)a | sol. (μM)a | LogDa | Hu PPB (% free)a | microsomal stabilityb | hERG IC50 (μM) |
|---|---|---|---|---|---|---|---|---|
| 7a | <3 | ND | 300 | <1 | 3.1 | 3 | 22 | >33 |
| 7b | <3 | 2 | 81 | 8 | 2.4 | 8 | 15 | 21 |
| 7c | <3 | ND | 67 | <1 | 3.0 | 4 | 56 | >33 |
| 7d | <3 | 3 | 21 | <1 | 3.1 | 2 | 39 | 19 |
| 7e | <3 | 2 | 57 | 5 | 2.3 | 13 | 36 | 16 |
| 7f | <3 | 2 | 12 | 7 | 2.8 | 9.6 | 19 | ND |
| 7g | <3 | 42 | ND | 18 | 2.1 | 15 | 50 | >33 |
| 7h | <3 | 6 | 300 | 7 | 2.1 | 12 | <4 | 19 |
| 7i | <3 | 33 | 600 | <1 | 2.9 | 2.4 | 47 | 11 |
| 7j | <3 | 8 | 410 | 5 | 3.0 | 4.3 | 31 | 16 |
| 7k | <3 | 3 | ND | 1 | 2.1 | 12 | <4 | ND |
| 7l | <3 | 10 | 110 | 52 | 0.8 | 21 | 20 | ND |
Mean value of two experiments. Deviations were within < ±25%.
Intrinsic clearance (CLint) determined from rat liver microsome incubations (μL/min/mg); ND = not determined.
An X-ray cocrystal structure determination of 7l with recombinant human CK2 at 2.2 Å resolution revealed the inhibitor to bind as expected with N2 of the pyrazolo[1,5-a]pyrimidine core and the C7 NH interacting with the hinge region of the ATP-binding pocket (Figure 2).12 As noted previously, critical to the high binding affinity of this inhibitor class is the engagement with a buried water molecule adjacent to the gatekeeper residue (Phe113). The energetically disfavored cisoid conformation adopted by the acetamide enables the carbonyl group to coordinate with the water molecule and with Lys68 and permits formation of a hydrogen bond between the acetamide NH and Asp175. The sum of these interactions, in addition to further coordination of the water molecule by the cyano group (3.0 Å) appears sufficient to compensate for the higher energy form of the acetamide. Deletion of the 3-cyano group from 7b to give des-cyano analogue 8 (not shown) results in a >100-fold reduction in enzymatic potency (CK2 IC50 = 0.27 μM) and loss of measurable cellular activity (pAKTS129 IC50 > 3.3 μM). A similar potency decrease was observed with the des-cyano analogue of indole 1a.8 In addition, we believe the increased potency of the anilino-compounds relative to 1a is due to steric reinforcement of the cisoid amide geometry by the ortho-substituent. Preorganization of the amide in a conformation required for recognition by the kinase presumably decreases the entropy loss upon binding of the ligand and results in higher affinity. This substitution may be essential for increased cellular activity as well; the growth inhibitory effects (HCT-116 GI50s) reported by Polaris for a series of related analogues lacking substitution ortho to the acetamide are 2–3 log units weaker than those in this study.13
Figure 2.
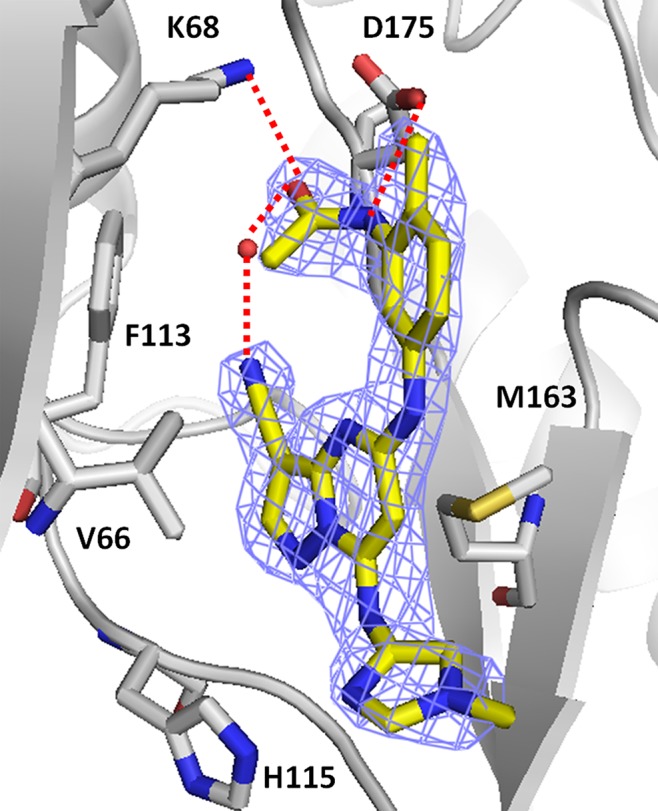
Human CK2 kinase in complex with 7l (PDB code: 4GUB) as determined at 2.2 Å resolution. The structure is oriented with the N-terminal lobe toward the top left, the C-terminal lobe toward the bottom right, and residues of the hinge region on the bottom left. The electron density (2Fo–Fc), contoured at 1σ, is shown as a wire mesh. Polar interactions ≤3.0 Å are indicated as dotted lines and bound water molecules are shown as red spheres.14
The N-methyl group of the imidazole in 7l is positioned at the interface with the solvent-accessible surface and suggests that further substitution at this position could be undertaken.
Through the intermediacy of hydroxyethyl derivatives 9a and 9b, we prepared a series of amine-containing analogues (Scheme 2). While our earlier attempts to modify indole 1a by introducing hydrophilic groups on the C7 substituent resulted in decreased cellular potency, we were initially encouraged to find that N-substituted analogues of 7l possessed high enzymatic and cellular activity (Table 2). In the pyrazole series (10a and 11a-c), these modifications resulted in a modest five- to 10-fold decrease in cellular potency (pAKTS129) yet maintained activity in the phenotypic assay in the sub-100 nM range. Despite possessing high aqueous solubility and low intrinsic clearance, the plasma concentrations of 11b–d following a single oral dose in the rat were extremely low (Cmax < 0.1 μM). This may be rationalized in part by considering the low measured permeability of 11b (Papp A–B = 0.2 × 10–6 cm/s) and high efflux (efflux ratio = 100) in MDR1 expressing LLCK cells. Similarly functionalized imidazoles (10b and 11d-f) exhibited a more significant drop off in cellular potency and were not profiled in vivo.
Scheme 2.

Table 2. Enzymatic/Cellular Activity and ADME/Physical Property Data for 10a,b and 11a–f.
| compd | CK2α IC50 (nM)a | HCT-116 GI50 (μM)a | sol. (μM)a | LogDa | CLintb |
|---|---|---|---|---|---|
| 10a | <3 | 0.08 | 22 | 1.8 | 17 |
| 10b | <3 | 0.04 | 83 | 1.6 | 43 |
| 11a | <3 | 0.07 | 860 | 2.3 | 80 |
| 11b | <3 | 0.10 | 3 | 2.5 | 65 |
| 11c | <3 | 0.97 | 10 | 1.3 | 31 |
| 11d | <3 | 0.65 | 84 | 1.5 | 25 |
| 11e | <3 | 0.65 | 110 | 1.7 | 30 |
| 11f | <3 | 0.67 | 120 | 1.4 | 63 |
Mean value of at least two experiments. Deviations were within < ±25%.
Intrinsic clearance determined from rat liver microsome incubations (μL/min/mg).
We next focused on a strategy for achieving further reduction in the lipophilicity of 7b and related compounds that would not rely on introduction of basic groups. In recent years, oxetanes have emerged in the synthetic and medicinal chemistry literature as a versatile functional group with the ability to modulate basicity, confer metabolic stability, and to reduce lipophilicity in drug-like molecules.15,16
A series of N7 3-oxetanyl analogues (12a–e) was prepared to compare the effect of this substitution on potency, physical properties, and ADME characteristics. Consistent with our expectations, in a series of matched molecular pairs, this modification lowered logD by ∼0.8 units, significantly decreased the fraction of compound bound by human plasma proteins, increased metabolic stability in rat liver microsome and hepatocyte incubations, and reduced activity toward the hERG ion channel (Table 3 and Figure 3). These compounds are potent inhibitors of CK2 (IC50 < 3 nM) but are 10-fold less potent in the mechanistic and phenotypic assays than their N7 cyclopropyl counterparts and, as a consequence, oxetane 12b possesses comparable activity to the isostructural N7 cyclobutyl derivative 7i (pAKT IC50 = 0.03 μM).
Table 3. Enzymatic/Cellular Activity and ADME/Physical Property Data for 12a–e.
| compd | CK2α IC50 (nM)a | pAKTS129 IC50 (nM)a | HCT-116 GI50 (μM)a | Sol. (μM)a | LogDa | Hu PPB (% free)a | CLintb | hERG IC50 (μM) |
|---|---|---|---|---|---|---|---|---|
| 12a | <3 | ND | 0.97 | 23 | 2.2 | 12 | <4 | ND |
| 12b | <3 | 28 | 0.28 | 13 | 1.6 | 28 | 17 | >33 |
| 12c | <3 | 21 | 0.61 | 22 | 1.5 | 26 | 6 | >33 |
| 12d | <3 | 6 | 0.059 | 11 | 2.2 | 20 | <4 | >33 |
| 12e | <3 | 3 | 0.074 | 8 | 2.1 | 21 | <4 | >33 |
Mean value of at least two experiments. Deviations were within < ±25%.
Intrinsic clearance determined from rat liver microsome incubations (μL/min/mg); ND = not determined.
Figure 3.

Relationship between lipophilicity (logD), human plasma protein binding (log Ki), and microsomal stability (CLint) and the N7 substituent in a series of matched molecular pairs. Markers are colored by the C5 substituent and arranged vertically according to N7 group.
However, when the N7 oxetane group is combined with an aniline bearing a cyclopropyl group ortho to the acetamide, as in 12d and 12e, cellular potency is improved, presumably due to an overall increase in lipophilicity. The pharmacokinetic profiles of 12d (F = 47%) and 7b (F = 25%) in the rat (10 mg/kg, PO) are characterized by good bioavailability and low clearance although the plasma half-life of 12d (t1/2 = 1.4 h) is decreased relative to that of 7b (t1/2 = 2.6 h) and may be attributed to increased in vivo clearance (CL = 23 mL/min/kg). Dissapointingly, the favorable PK characteriristics of 12d were not shared by other analogues in this series; oxetanes 12b and 12e exhibit reduced oral bioavailability (F < 5%) and plasma levels (Cmax < 1 μM). These results may be explained, at least in part, by considering the aqueous solubility data for 12a–e, which show little, if any, enhancement relative to the cyclopropyl matched pairs.
Kinase selectivity profiling of 7b against a panel of 402 kinases revealed a high degree of selectivity (Figure 4).17 Limited off-target activity (10 kinases with >40% inhibition following treatment with 7b at a concentration of 0.1 μM/L) was observed against members of the CMGC family, including isoforms of the dual-specificity tyrosine-regulated kinases (Dyrk) and CAMK kinases such as the death-associated protein kinase (Dapk) and histone interacting protein kinase (Hipk) families. Biochemical IC50 determinations revealed an activity pattern similar to that of indole 1a from our earlier work with 7b exhibiting submicromolar activity against Hipk2 (IC50 = 40 nM), Dyrk3 (IC50 = 70 nM), and PIM3 (IC50 = 150 nM) with weaker effects on GSK3β (IC50 = 2.6 μM) and CDK2 (IC50 = 7.2 μM).
Figure 4.
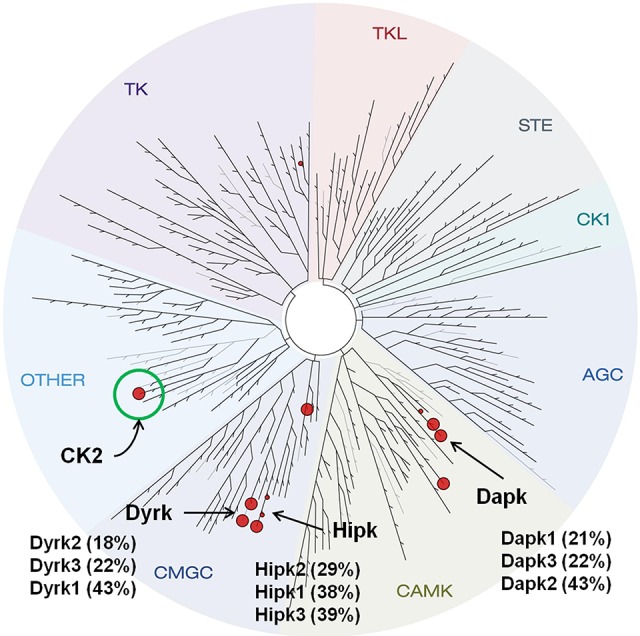
Kinome-wide selectivity profile of 7b. Red spheres highlight kinases for which the residual activity remaining after treatment with the inhibitor at a concentration of 0.1 μM/L was less than 45% of DMSO control using a competition binding assay that measures the ability of a compound to compete with an immobilized, active-site directed ligand (KINOMEscan, Discoverx, Inc., San Diego, CA).18
On the basis of their physical properties, pharmacokinetic profiles in rodents and dog, and potency against a range of cancer cell lines, 7b and 7e were chosen as probes to assess the potential of the series in pharmacodynamic (PD) and disease model studies. Treatment of murine DLD-1 AKT1 overexpressing xenografts with a single dose of 7b (10 mg/kg, PO) results in inhibition of pAKTS129 at the 3 and 8 h time points, but the effect is not sustained at 24 h. Treatment with 7e (12.5 mg/kg, PO) also depletes pAKTS129 at the early time points and appears to have a partial effect at 24 h as determined by Western blot analysis (Figure 5). Dose/response plots illustrating pAKTS129 inhibition following treatment with 7b, as measured by ELISA of tumor cell lysates, indicate residual inhibition of pAKTS129 at later time points, when no detectable drug remains in plasma. Compound 7b showed limited tumor growth inhibition when tested using a once daily 10 mg/kg oral dose (TGI = 28%) or a twice daily 5 mg/kg oral dose (TGI = 37%) for 14 days in disease model studies using a murine DLD-1 xenograft.
Figure 5.
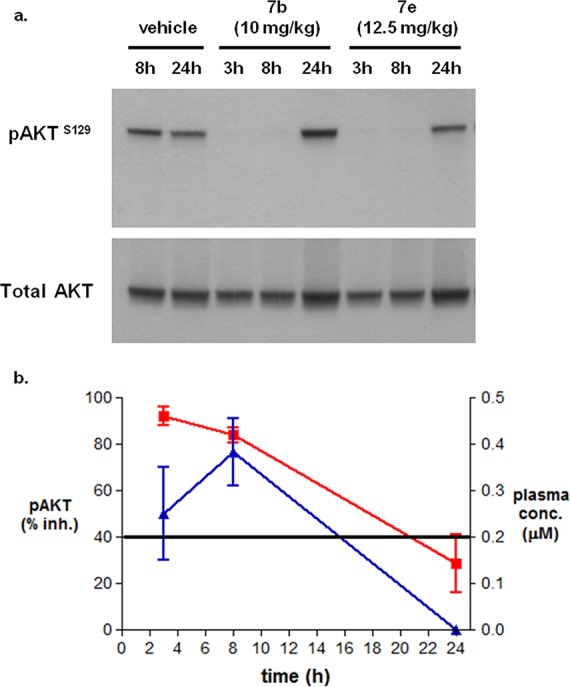
(a) Western blot analysis of pAKTS129 levels in tumor cell lysates following administration of 7b (10 mg/kg, PO) and 7e (12.5 mg/kg, PO) in a DLD-1 AKT1 overexpressing murine xenograft model. (b) Dose/response plot illustrating pAKTS129 levels (red) in tumor cell lysates, as determined by ELISA, versus plasma concentration of 7b (blue).
In particular, we have demonstrated that 3-amino oxetane derivatives exhibit reduced lipophilicity, decreased affinity for plasma proteins, and reduced hERG ion channel binding. Additional studies to determine the relationship between exposure and pharmacodynamic response for these and related agents in vivo will be described in subsequent communications.19
Supporting Information Available
Representative experimental procedures for synthesis of analogues, biochemical and cellular assays, and X-ray structure determination. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Ruzzene M.; Pinna L. A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells?. Biochim. Biophys. Acta 2010, 1804, 499–504. [DOI] [PubMed] [Google Scholar]
- Di Maira G.; Brustolon F.; Pinna L. A.; Ruzzene M. Dephosphorylation and inactivation of Akt/PKB is counteracted by protein kinase CK2 in HEK 293T cells. Cell. Mol. Life Sci. 2009, 66, 3363–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez I.; Sonenshein G. E.; Seldin D. C. CK2 and its role in Wnt and NF-kappa B signaling: Linking development and cancer. Cell. Mol. Life. Sci. 2009, 66, 1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laramas M.; Pasquier D.; Filhol O.; Ringeisen F.; Descotes J.-L.; Cochet C. Eur. J. Cancer 2007, 43, 928–934. [DOI] [PubMed] [Google Scholar]
- Ji H.; Wang J.; Nika, Trembley J. H.; Unger G. M.; Tobolt D. K.; Korman V. L.; Wang G.; Ahmad K. A.; Slaton J. W.; Kren B. T.; Ahmed K. Systemic administration of antisense oligonucleotides simultaneously targeting CK2alpha and alpha’ subunits reduces orthotopic xenograft prostate tumors in mice. Mol. Cell. Biochem. 2011, 356, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarno S.; Papinutto E.; Franchin C.; Bain J.; Elliott M.; Meggio F.; Kazimierczuk Z.; Orzeszko A.; Zanotti G.; Battistutta R.; Pinna L. A. ATP site-directed inhibitors of protein kinase CK2: an update. Curr. Top. Med. Chem. 2011, 11, 1340–1351. [DOI] [PubMed] [Google Scholar]
- Pierre F.; Chua P. C.; O’Brien S. E.; Siddiqui-Jain A.; Bourbon P.; Haddach M.; Michaux J.; Nagasawa J.; Schwaebe M. K.; Stefan E.; Vialettes A.; Whitten J. P.; Chen T. K.; Darjania L.; Stansfield R.; Anderes K.; Bliesath J.; Drygin D.; Ho C.; Omori M.; Proffitt C.; Streiner N.; Trent K.; Rice W. G.; Ryckman D. M. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c] [2,6] naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54, 635–54. [DOI] [PubMed] [Google Scholar]
- Dowling J. E.; Bao L.; Brassil P.; Chen H.; Chuaqui C.; Cooke E. L.; Denz C. R.; Larsen N. A.; Lyne P. D.; Peng B.; Pontz T. W.; Racicot V.; Russell D.; Su N.; Thakur K.; Wu A.; Ye Q.; Zhang T. Potent and selective inhibitors of CK2 kinase identified through structure-guided hybridization. ACS Med. Chem. Lett. 2012, 3, 278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maira G.; Brustolon F.; Pinna L. A.; Ruzzene M. Dephosphorylation and inactivation of Akt/PKB is counteracted by protein kinase CK2 in HEK 293T cells. Cell. Mol. Life Sci. 2009, 66, 3363–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranenburg M.; van der Burgt Y. E. M.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Goubitz K.; Fraanje J. New diphosphine ligands based on heterocyclic aromatics inducing very high regioselectivity in rhodium-catalyzed hydroformylation: effect of the bite angle. Organometallics 1995, 14, 3081–3089. [Google Scholar]
- See Supporting Information.
- PDB deposition code for the cocrystal structure is 4GUB.
- Nie Z.; Perretta C.; Erickson P.; Margosiak S.; Almassy R.; Lu J.; Averill A.; Yager K. M.; Chu S. Structure-based design, synthesis, and study of pyrazolo[1,5-a][1,3,5]triazine derivatives as potent inhibitors of protein kinase CK2. Bioorg. Med. Chem. Lett. 2007, 17, 4191–4195. [DOI] [PubMed] [Google Scholar]
- Figure produced using Pymol. DeLano W. L.The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, 2002; see http://www.pymol.org. [Google Scholar]
- Wuitschik G.; Carreira E. M.; Wagner B.; Fischer H.; Parrilla I.; Schuler F.; Rogers-Evans M.; Müller K. Oxetanes in drug discovery: Structural and synthetic insights. J. Med. Chem. 2010, 5383227–3246. [DOI] [PubMed] [Google Scholar]
- Burkhard J. A.; Wuitschik G.; Rogers-Evans M.; Müller K.; Carreira E. M. Oxetanes as versatile elements in drug discovery and synthesis. Angew. Chem., Int. Ed. 2010, 49489052–9067. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Image generated using TREEspot Software Tool and reprinted with permission from KINOMEscan, a division of DiscoveRx Corporation, © DISCOVERX CORPORATION 2010.
- Dowling J. E.; Alimzhanov M.; Bao L.; Chuaqui C.; Denz C.; Ferguson A.; Graff C.; Liu Z.-Y.; Lyne P.; Racicot V.; Wu A.; Wu J.; Ye Q.; Cooke E. L.. Discovery and characterization of AZ968, a potent and selective inhibitor of CK2 kinase with effects on AKT signaling in vivo. Poster presented at the 103rd Annual Meeting of the American Association for Cancer Research, March 31–April 4, 2012. Chicago, IL; Abstract No. 3907.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.