

Abstract
Here, we describe the selection and optimization of a chemical series active in both a full-length and a fragment-based Huntington’s disease (HD) assay. Twenty-four thousand small molecules were screened in a phenotypic HD assay, identifying a series of compounds bearing a 3-hydroxy-3-trifluoromethylpyrazole moiety as able to revert the toxicity induced by full-length mutant Htt by up to 50%. A chemical exploration around the series led to the identification of compound 4f, which demonstrated to be active in a Htt171–82Q rat primary striatal neuron assay and a PC12-Exon-1 based assay. This compound was selected for testing in R6/2 mice, in which it was well-tolerated and showed a positive effect on body weight and a positive trend in preventing ventricular volume enlargment. These studies provide strong rationale for further testing the potential benefits of 3-hydroxy-3-trifluoromethylpyrazoles in treating HD.
Keywords: Huntington’s disease, mutant Htt toxicity, phenotypic screening, 3-hydroxy-3-trifluoromethylpyrazoles, ADME, R6/2 mouse model
Huntington’s disease (HD) is an autosomal-dominant neurodegenerative brain disorder caused by a CAG trinucleotide repeat expansion (>35 repeats), which encodes an abnormally long polyglutamine (polyQ) tract in the N-terminal part of a large protein called huntingtin (Htt).1 Mutant Htt is subject to cleavage by proteolytic enzymes1 and to aberrant pre-RNA splicing,2 which results in the generation of N-terminal fragments containing the expanded polyQ.1 These fragments are able to aggregate with themselves and other proteins and form large nuclear and cytoplasmatic inclusions.3 Although the scientific community is still debating the role of these aggregates, the affected neurons suffer from many different dysfunctions altering the normal cell equilibrium and leading to apoptotic or necrotic cell death.4 Among these dysfunctions, reduced ATP levels, decreased Ca2+ uptake, oxidative stress, excitotoxicity, altered transcription, and impaired autophagy are reported as major pathogenic mechanisms.5 Taken together, the pathophysiological picture of HD still remains very complex and poorly understood, lacking validated target proteins for therapeutic intervention. Furthermore, despite significant research efforts in the last 20 years since the mutated huntingtin gene was discovered,6 none of the drug discovery programs aimed at tackling a single target or a single pathway involved in the disease pathology has yet yielded a molecule capable of interfering with disease progression.7
Current target-based drug discovery strategies for HD are of limited use, and alternative approaches for which an a priori knowledge of molecular targets is not necessary are desirable. Toward this goal, several groups have developed phenotypic strategies to identify small molecules to be further developed into potential drug candidates (Figure 1).8 The antiaggregation activity of the Rho-associated protein kinase 1 (Rock1) inhibitor Y-27632 1, identified in a HEK-293 cell system, was subsequently demonstrated to be active in HD-relevant cell and animal models.9 Compounds 2 and 3 were identified in cell screenings respectively as selective enhancer of mutant Htt clearance10 and active in a cell viability assay against HD-induced neurodegeneration.11 Both compounds resulted also active in follow-up animal models, including mouse and C.elegans models of HD.12,13
Figure 1.
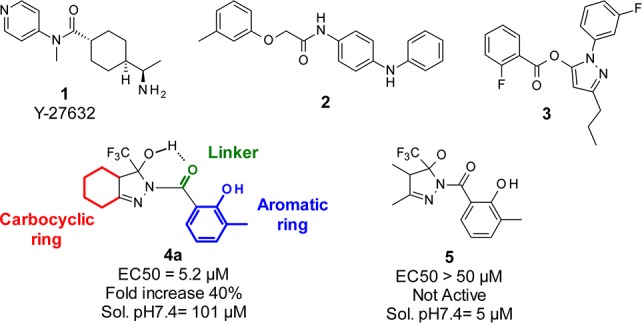
Chemical structures of hit compounds identified in HTS HD screenings.
Our approach aimed at identifying a class of compounds displaying activity in both full-length and Exon-1 mutant huntingtin-based HD assays, thus enabling us to recapitulate the animal models we planned to use for preclinical compound profiling (R6/2, Exon-1 based) and the human version of the disease. Although not exhaustive, we sought to build a paradigm to maximize the chance for effective translation of preclinical results toward clinical trials (Figure 2).
Figure 2.
General workflow.
An HTS method was developed in-house creating a stable recombinant 293/T-Rex cell line generated with both a CRE-luciferase (CRE-LUC) reporter gene and with the full-length mutant Htt gene under control of an inducible CMV promoter; it has been shown that mutated Htt sequesters the cAMP response element-binding protein (CREB) coactivator, CREB-binding protein (CBP) through direct protein interactions, which leads to decreased CREB-mediated transcription.14 In addition to this, we planned to use another in vitro model of HD based on Htt expression via LV infection on primary striatal rat neurons as a secondary screening assay. This assay relies on the incorporation of a Htt-derived sequence expressing an N-terminal 171 aa fragment of mutant or wild-type Htt (Htt171–82Q or Htt171–18Q, respectively; see Supporting Information).15
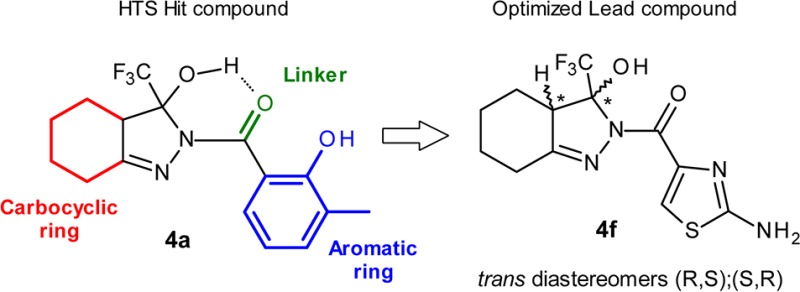
For the HTS screening campaign we selected 24,000 small organic molecules from the diverse Siena Biotech compound collection. Among the most promising hit compounds, a small set of molecules containing a fused 3-hydroxy-3-trifluoromethylpyrazole moiety, initially consisting of 4 compounds and exemplified by compound 4a, displayed an activity range between 5.9 and 18 μM with fold increase (FI) values between 30% and 50% as a measure of efficiency of the compound to restore the CREB-mediated transcriptional activity in cells expressing mutant Htt. A set of nonfused analogues represented by compound 5 proved inactive in the screening when tested up to 50 μM, showing the selectivity of this specific chemotype only when fused to a cyclic ring.
A major concern of this series was the presence of the geminal 3-hydroxy-3-trifluoromethyl functionality and its stability to dehydration. Indeed, it is reported in the literature that 2-aryl or 2-alkyl substituted 3-hydroxy-3-trifluoromethyl hexahydroindazoles undergo dehydration in acidic conditions to afford the corresponding 3-trifluoromethyl tetrahydroindazoles derivatives.16 After retest from a new batch and a preliminary stability test conducted at pH = 7.4 and pH = 3, the 2-acyl and 2-sulphonyl hexahydroindazoles confirmed activity and stability to dehydration (data not shown). We speculate that in this particular assembly the carbonyl oxygen atom could stabilize the 3-hydroxyl group on the pyrazole ring from dehydration by an intramolecular hydrogen bond interaction (see compound 4a in Figure 1).
In the optimization program, we opted for maintaining the main structural features of the molecules in order to keep the general pharmacophore shape and focused on the exploration of three main points: (a) the carbocyclic ring, (b) the linker, and (c) the R1 ring (see Figure 1).
Initial hit 4a showed acceptable solubility and permeability, but a far too high metabolism rate in human and mouse. In an effort to improve the overall profile of 4a, mitigating its metabolic stability and moving to a IP-free chemical space, we decided to explore the insertion of different heterocycle rings in R1 position, and few analogues were synthesized (see Scheme 1). The presence of an heterocycle in R1 not only produced a general improvement in the metabolic stability of the compounds but also favored solubility and permeability across the series (see Table 1).
Scheme 1. General Synthetic Route for the Fused 3-Hydroxy-3-trifluoromethylpyrazole Derivatives.

Reagents and conditions: (a) ethyl trifluoroacetate, NaOMe, Et2O, −10 °C to RT; (b) glacial AcOH; (c) acylhydrazide, pyrrolidine, mol sieves, 0 °C to RT, THF; (d) sulphonylhydrazide, pyrrolidine, mol sieves, 0 °C to RT, THF.
Table 1. CRE-LUC Activity and ADME Properties of Fused 3-Hydroxy-3-trifluoromethylpyrazole Derivatives.

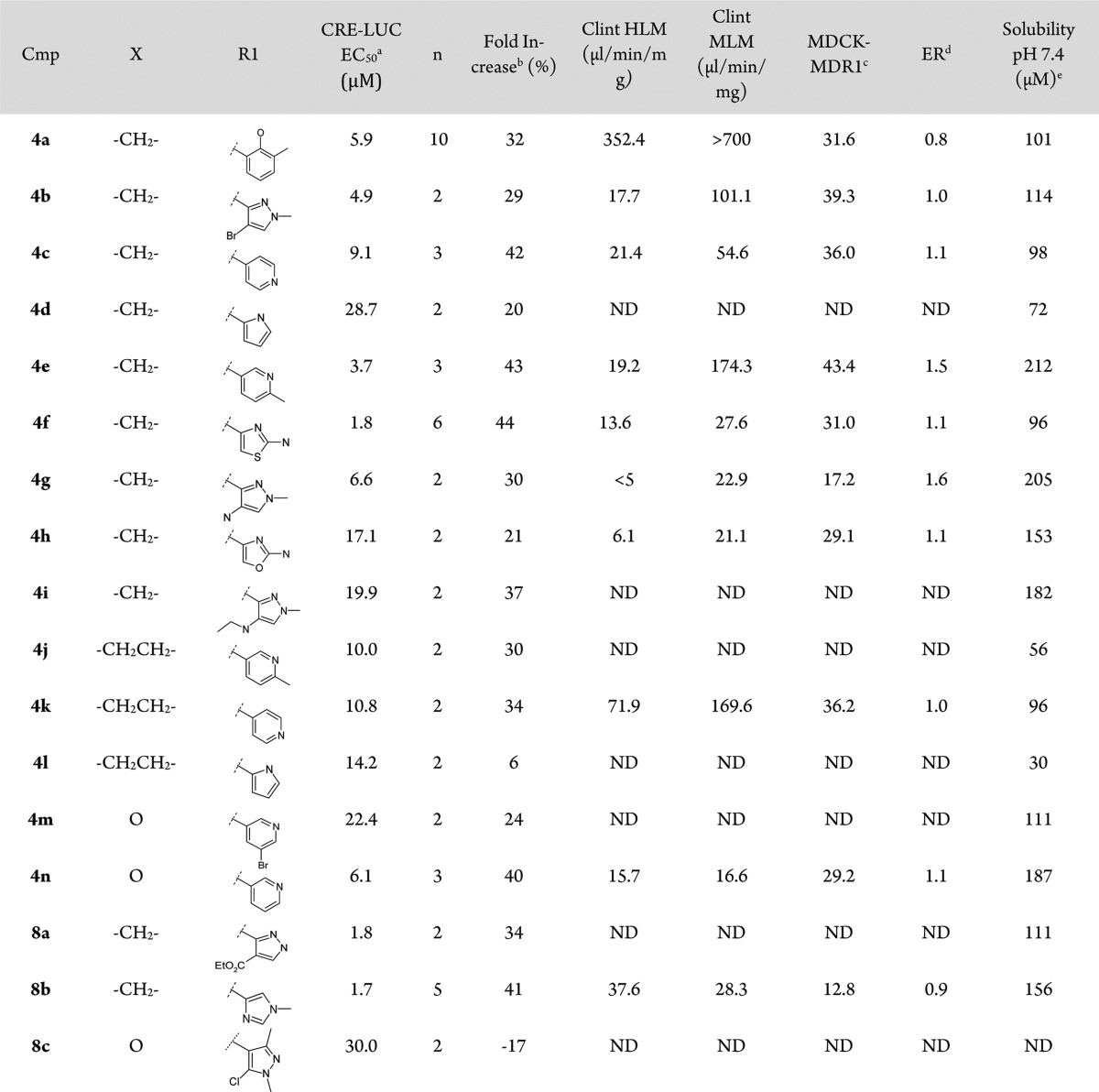
Cellular assay. EC50 values represent arithmetic means of n reported determinations. These assay generally produced results within 2-fold of the reported mean. Compound 1 (Y-27632) was used as a standard and positive control for the assay.
Fold increase is considered as the measure of the difference of maximum efficacy calculated from the concentration response curve fit of the normalized CRE-LUC activity of the mutant Htt expressing cells (Emax-ind) and of the noninduced cells (Emax-nonind).
Permeability assay A:B (10–6 cm/s).
Efflux ratio measured in MDCK-MDR1 permeability assay (B:A/A:B).
Kinetic solubility.
As shown in Scheme 1, the fused 3-hydroxy-3-trifluoromethylpyrazoles 4 and 8 were prepared starting from the corresponding cyclic ketones 6. Various cyclic rings were explored starting from commercially available cyclohexanone, cycloheptanone, and 4-pyranone. Upon treatment with sodium methoxide and ethyl trifluoroacetate in diethyl ether, the diketone 7 was isolated as the corresponding sodium salt, or as a mixture in the keto–enol form. Cyclization of the diketone with a substituted acylhydrazide or sulphonylhydrazide in the presence of pyrrolidine yielded exclusively the 2-substituted acyl- or sulphonyl-hexahydroindazoles with yields ranging 10–70% and no trace of the N1-substituted isomer.
Noncommercially available acylhydrazides 11 and sulphonylhydrazides 13 (Scheme 2) were generally obtained following a published procedure17 from corresponding carboxylic acids 9 by transformation into the methyl ester 10 and treatment with hydrazine monohydrate;18,19 sulphonylhydrazides 13 were obtained from corresponding sulphonyl chlorides 12 by treatment with hydrazine monohydrate.20,21 See Supporting Information for details.
Scheme 2. General Synthetic Route for Acylhydrazide and Sulphonylhydrazide Derivatives.

Reagents and conditions: (a) thionyl chloride, MeOH, 0 °C; (b) hydrazine monohydrate, 50 °C; (c) hydrazine monohydrate, THF, 0 °C.
The stereospecificity of the reaction was confirmed by X-ray diffraction data obtained for compound 4f (see Supporting Information for full data and 3D representations). The data confirmed the presence of a racemate having the −H and −OH substituents, respectively, on the C1 and C2 chiral centers trans to each other as a mixture of (R,S) and (S,R).22 No cis diastereomers analogues were consistently detected. Carbocyclic ring expansion to a 7-membered ring did not improve potency, with compounds 4j–4l showing double digit EC50s and fold-increase values below 30%.
Furthermore, cycloheptane derivatives showed increased metabolism rates, such as for compound 4j, mainly due to carbocyclic chain hydroxylation (data not shown). No improvements were detected with the insertion of a pyrane ring, with compound 4n showing comparable cellular potency to initial hit 4a. The best results in terms of potency and efficiency were obtained with acyl derivatives 4f and sulphonyl derivative 8b exibiting a 6-membered carbocyclic ring and a 5-membered heterocycle in R1 position as the most favorable combination with both linkers. Our phenotypic assay approach has thus lead to the selection of a subseries of molecules that showed FI and EC50 values feasible for further evaluation in the primary rat striatal neuron secondary assay.
Compounds 4b, 4c, 4f, and 8b were tested at EPFL in Lausanne at 1, 10, and 30 μM. In this primary rat striatal neuron-based model, Htt171–18Q or Htt171–82Q expression is driven by a tetracycline responsive element (TRE) promoter (TRE-htt171–18Q/82Q) in a lentiviral expression vector, producing a rapid polyQ-dependent pathology that leads to formation of intracellular Htt inclusions starting at 1–2 weeks and decreased expression of the neuronal marker NeuN (a readout of viability) as early as 2 weeks after infection. All four compounds were shown to significantly improve NeuN counts relative to vehicle (DMSO-treated) counterparts at 10 and 30 μM concentrations (see Figure S1 in Supporting Information). Figure 3A shows the effect on survival of compound 4f as representative of the series. The effect was not completely specific, with an increased NeuN count also detectable in the htt171–18Q expressing cells. Our deduction was that the potential target(s) of this series of compounds is involved in a general cell viability pathway. To further build upon this result, the cytoprotection of compound 4f was tested in a PC12 cell model in which Exon-1 mut-Htt expression is under control of a tetracycline (Tet-on) system.23 Again, the compound was able to protect from doxycycline (Doxy)-induced cell death by reducing LDH release, a readout of mortality (Figure 3B). In this model no effects of the compound were evident in uninduced cells.
Figure 3.
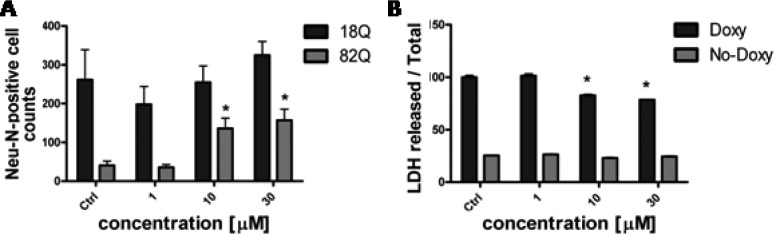
(A) Effects of 4f treatment on LV-Htt (wild-type htt171–18Q and mutant htt171–82Q) infected striatal neurons. *p < 0.05 Student’s t test vs 82Q Ctrl. (B) Protection against cell death in Doxy-induced Exon 1-mutHTT expressing cells (Doxy). *p < 0.05 vs Doxy-induced Ctrl group (Two-Way ANOVA, Tukey’s posthoc test). Data are expressed as mean and SEM values.
More generally, all compounds examined did not show any toxicity on normal cells (or neurons) even for a long period of treatment (results not shown). Potential off-target effects of the series were evaluated in an early representative, compound 4b, which was tested in a CEREP panel. The CEREP express screening was performed on 71 receptors and 62 enzymes in which 4b was profiled at 20 μM concentration. Compound 4b showed no significant activity on any target (full list and result values available in Supporting Information) providing us with enough information and confidence to consider the series potentially free from most common off-target effect and ready to enter into animal studies.
Following the achievement of a proof of activity in a full-length, an Exon-1-based and an N171-based HD cell model, it was decided to progress investigation of the series in an in vivo model of HD. Selection criteria were based on compounds with the best achievable systemic exposure in the animal. Selected compounds were progressed to PK experiments (see Table S1 in Supporting Information) to determine whether a suitable candidate for animal model testing (R6/2 mice) could be identified. All analyzed compounds showed very short half-lives with no remaining compound detected at 24 h after administration. The Sulphone derivative 8b showed a high clearance in mouse and very low levels after PO administration, while the carbonyl derivatives 4f and 4c showed medium to optimum bioavailability values (see Table S1 in Supporting Information). Compound 4f was selected as a lead compound after a dose escalating PK and an MTD experiment in which it showed suitable systemic exposure and tolerability properties in mouse (data not shown) making it compatible with a chronic administration protocol. Additional studies on 4f determined a B/P ratio of 0.34, no hERG activity detected up to 100 μM concentration, negative mini-Ames test, and no CYP inhibition (3A4 TST > 30 μM; 3A4 NIF > 30 μM; 1A2 > 30 μM; 2D6 > 30 μM; 2C9 > 30 μM; 2C19 = 24.4 μM).
R6/2 mice were treated with 4f at 3, 10, and 30 mg/kg, PO, QD at Psychogenics (Tarrytown, NY), starting from 4 weeks of age. Effects of compound treatment were assessed with a motor test battery comprising open field, grip strength, and rotarod. Drug effects were also evaluated on BW and followed-up with survival assessment. Compound 4f showed a significant effect on the clinically relevant parameter of BW, counteracting the characteristic decrease in BW at doses of 10 and 30 mg/kg (see Figure S3 in Supporting Information). No significant effects of treatment were detected on motor tests or survival.
A small satellite group of animals was sacrificed at 12 weeks of age (after 8 weeks of treatment) to perform brain morphological studies. Effects of 4f was assessed on the neuropathological hallmarks of brain atrophy and ventricular enlargement, using both ex vivo MRI and histological measures. The quantification of EM48-positive nuclear Htt aggregates were also analyzed by immuno-histochemistry. Results of morphological studies (Figure 4) showed a severe enlargement of cerebral ventricular in R6/2 animals as compared to the wild-type strain (C57Bl6). Furthermore, compound 4f showed a trend toward decreasing the size of the cerebral ventricles. These results were also confirmed by ex vivo MRI performed in fixed brain (see Figure S3 in Supporting Information).
Figure 4.
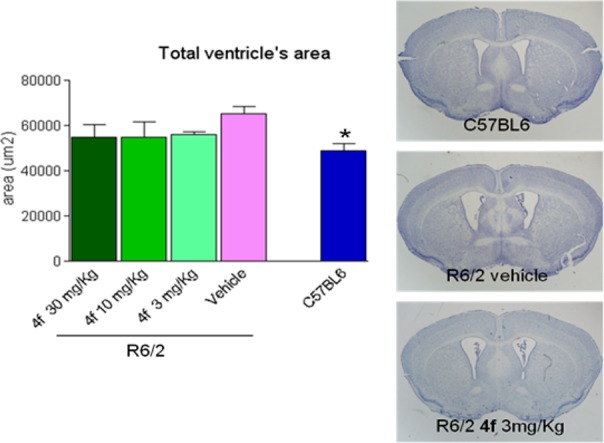
Effects of 4f treatment at several doses on brain ventricular enlargement in R6/2 animals. Brain photomicrographs as exemplum of analyzed slices; histogram, quantitative analysis. *p < 0.05 vs R6/2 vehicle (One-Way ANOVA, Tukey’s posthoc test).
In conclusion, we have identified a class of compounds active in three different HD cell assays comprising different phenotypes, readouts, and promoters of toxicity induced by mutant Htt. Overall, the class showed favorable ADME properties and in particular compound 4f showed a good profile that was judged suitable for in vivo studies. Results of R6/2 mouse testing showed a significant effect on body weight and a positive trend in decreasing cerebral ventricle volume, but no effect on motor tests or survival. Although our compounds showed good activity in various in vitro HD models, the modest findings of a statistically positive BW response and a trend in improving ventricular volume did not provide a sufficiently strong impetus for us to develop the compound further. To fully understand the potential of this class of compounds, additional studies are warranted. A target deconvolution strategy could be of particular help to understand the potential of the newly identified series either by improving potency/efficacy or by identifying the target or inferring the molecular pathway in which this class of molecules is involved.24
Acknowledgments
We thank Dr. Francesco Berrettini (Università degli Studi di Siena) for the X-ray diffraction data collection and analysis; Dr. Carol Murphy and Dr. Sylvie Ramboz at Psychogenics, Tarrytown, NY, for in life phase of R6/2 experiments.
Glossary
Abbreviations
- Htt
Huntingtin
- CRE
cAMP response element
- BW
body weight
- HTS
high throughput screening
- LDH
lactate dehydrogenase
- B/P
brain/plasma
Supporting Information Available
Synthetic procedures and characterization data of compounds, X-ray crystallography data for compound 4f, CEREP selectivity data for compound 4b, PK data, biological assay procedures, and R6/2 mouse model methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ (S.L.R.) Children’s Tumor Foundation, 95 Pine Street, 16th Floor, New York, New York 10005-1703, United States.
Author Present Address
∥ (F.H.) GenKyotex SA, Geneva, Switzerland.
Author Present Address
⊥ (R.L.-C.) Department of Cell Physiology and Pharmacology, University of Leicester, Leicester, United Kingdom.
Author Present Address
○ (V.M.) Department of Veterinary Sciences, University of Pisa, Viale delle Piagge 2, 56124 Pisa, Italy.
The authors declare no competing financial interest.
Supplementary Material
References
- Tobin A. J.; Signer E. R. Huntington’s disease: the challenge for cell biologists. Trends Cell Biol. 2000, 10, 531–536. [DOI] [PubMed] [Google Scholar]
- Sathasivam K.; Neueder A.; Gipson T. A.; Landles C.; Benjamin A. C.; Bondulich M. K.; Smith D. L.; Faull R. L. M.; Roos R. A. C.; Howland D.; Detloff P. J.; Housman D. E.; Bates G. P. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M.; Finkbeiner S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 23811–11. [DOI] [PMC free article] [PubMed] [Google Scholar]; Arrasate M.; Finkbeiner S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 238, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bano D.; Zanetti F.; Mende Y.; Nicotera P. Neurodegenerative processes in Huntington’s disease. Cell Death Dis. 2011, 2, e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L., Hughes A.. Pathogenic Mechanisms in Huntington’s Disease. In Pathophysiology, Pharmacology, and Biochemistry of Dyskinesia; Brotchie J., Bezard E., Jenner P., Eds.; Academic Press: New York, 2011; Vol. 98, pp 373–418. [DOI] [PubMed] [Google Scholar]
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- Venuto C. S.; McGarry A.; Ma Q.; Kieburtz K. Pharmacologic approaches to the treatment of Huntington’s disease. Mov. Disord. 2012, 27, 31–41. [DOI] [PubMed] [Google Scholar]
- Heitz F.; La Rosa S.; Gonzalez-Couto E.; Gaviraghi G.; Terstappen G. C. Drug discovery and development for Huntington’s disease: an orphan indication with high medical need. Drugs 2008, 11, 653–660. [PubMed] [Google Scholar]
- Li M.; Huang Y.; Ma A. A. K.; Lin E.; Diamond M. I. Y-27632 improves rotarod performance and reduces huntingtin levels in R6/2 mice. Neurobiol. Dis. 2009, 363413–420. [DOI] [PubMed] [Google Scholar]
- Coufal M.; Maxwell M. M.; Russel D. E.; Amore A. M.; Altmann S. M.; Hollingsworth Z. R.; Young A. B.; Housman D. E.; Kazantsev A. G. Discovery of a novel small-molecule targeting selective clearance of mutant huntingtin fragments. J Biomol. Screening 2007, 12, 351–360. [DOI] [PubMed] [Google Scholar]
- Varma H.; Voisine C.; DeMarco C. T.; Cattaneo E.; Lo D. C.; Hart A. C.; Stockwell B. R. Selective inhibitors of death in mutant huntingtin cells. Nat. Chem. Biol. 2007, 3, 99–100. [DOI] [PubMed] [Google Scholar]
- Yamamoto A.; Lucas J. J.; Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000, 101, 57–66. [DOI] [PubMed] [Google Scholar]
- Varma H.; Lo D. C.; Stockwell B. R. High throughput screening for neurodegeneration and complex disease phenotypes. Comb. Chem. High Throughput Screening 2008, 11, 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzeroni G.; Benicchi T.; Heitz F.; Magnoni L.; Diamanti D.; Rossini L.; Massai L.; Federico C.; Fecke W.; Caricasole A.; La Rosa S.; Porcari V. A phenotypic screening assay for modulators of Huntingtin induced transcriptional dysregulation. J. Biomol. Screen. 2013, 10.1177/1087057113484802. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R.; Taylor D. M.; Pallos J.; Lambert E.; Amore A.; Parker A.; Moffitt H.; Smith D. L.; Runne H.; Gokce O.; Kuhn A.; Xiang Z.; Maxwell M. M.; Reeves S. A.; Bates G. P.; Neri C.; Thompson L. M.; Marsh J. L.; Kazantsev A. G. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 7927–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyga J. W.; Henrie R. N.; Meier G. A.; Creekmore R. W.; Patera R. M. ‘Through-space’ Hydrogen–Fluorine, Carbon–Fluorine and Fluorine–Fluorine spin–spin coupling in 2-phenyl-3-alkyl-4,5,6,7-tetrahydroindazoles. Magn. Reson. Chem. 1993, 31, 323–328. [Google Scholar]
- Friedman L.; Litle R. L.; Reichle W. R.. Organic Syntheses; Wiley: New York, 1973; Vol. 5, p 1055. [Google Scholar]
- Zhao H.; Burke T. R. Jr. Pentafluorophenyl ester activation for the preparation of N,N′-diaroylhydrazines. Tetrahedron 1997, 53, 4219–4230. [Google Scholar]
- Issartel V.; Spehner V.; Coudert P.; Seilles E.; Couquelet J. Synthesis of new pyrrolo[1,2-d][1,2,4]triazines and thiazolo[3,4-d][1,2,4]triazines as immunostimulating agents. Bioorg. Med. Chem. 1998, 6, 349–354. [DOI] [PubMed] [Google Scholar]
- House H. O.; Gaa P. C.; VanDerveer D. Perhydroazulenes. 3. Conformations of the 4-oxoperhydroazulenes. J. Org. Chem. 1983, 48, 1661–1670. [Google Scholar]
- Koide Y.; Uemoto K.; Hasegawa T.; Sada T.; Murakami A.; Takasugi H.; Sakurai A.; Mochizuki N.; Takahashi A.; Nishida A. Pharmacophore-based design of sphingosine 1-phosphate-3 receptor antagonists that include a 3,4-dialkoxybenzophenone scaffold. J. Med. Chem. 2007, 50, 442–454. [DOI] [PubMed] [Google Scholar]
- Teichert J.; Oulie P.; Jacob K.; Vendier L.; Etienne M.; Claramunt R. M.; Lopez C.; Perez Medina C.; Alkorta I.; Elguero J. The structure of fluorinated indazoles: the effect of the replacement of a H by a F atom on the supramolecular structure of NH-indazoles. New J. Chem. 2007, 31, 936–946. [Google Scholar]
- Wyttenbach A.; Swartz J.; Kita H.; Thykjaer T.; Carmichael J.; Bradley J.; Brown R.; Maxwell M.; Schapira A.; Orntoft T. F.; Kato K.; Rubinsztein D. C. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 1829–1845. [DOI] [PubMed] [Google Scholar]
- Terstappen G. C.; Schlüpen C.; Raggiaschi R.; Gaviraghi G. Target deconvolution strategies in drug discovery. Nat. Rev. Drug Discovery 2007, 11, 891–903. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.