

Abstract

Tetrahydropyrazolo[1,5-a]pyrimidine scaffold was identified as a hit series from a Mycobacterium tuberculosis (Mtb) whole cell high through-put screening (HTS) campaign. A series of derivatives of this class were synthesized to evaluate their structure–activity relationship (SAR) and structure–property relationship (SPR). Compound 9 had a promising in vivo DMPK profile in mouse and exhibited potent in vivo activity in a mouse efficacy model, achieving a reduction of 3.5 log CFU of Mtb after oral administration to infected mice once a day at 100 mg/kg for 28 days. Thus, compound 9 is a potential candidate for inclusion in combination therapies for both drug-sensitive and drug-resistant TB.
Keywords: Antituberculosis; tetrahydropyrazolo[1,5-a]pyrimidine; structure−activity relationship; structure−property relationship
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is an airborne infectious disease that infects almost one-third of the world’s human population.1 Effective chemotherapy for TB has existed since the 1940s; however, an important factor contributing to the resurgence of the disease is the emergence of multidrug resistant tuberculosis (MDR TB). MDR TB is caused by Mtb that is resistant at least to isoniazid and rifampicin, the two most potent anti-TB drugs. Current treatment for MDR TB consists of lengthy regimens that are less effective, more expensive, and less well tolerated.2 Therefore, there remains an urgent need for new TB drugs that are able not only to shorten the long treatment regimen but also to control drug resistant forms of TB and that can be used along with the current AIDS/HIV retroviral treatments.3
To identify a new starting point in the development of new TB drugs, the Novartis internal small molecule chemical library was screened for activity against Mycobacterium bovis BCG as a surrogate of Mtb by measuring ATP levels using the BacTiter-Glo assay as described earlier.4 Subsequent hit confirmation with Mtb H37Rv led to the identification of tetrahydropyrazolo[1,5-a]pyrimidine scaffold as one of the hit series. Two other groups have also independently reported this scaffold as a hit from their own phenotypic high-throughput screening campaigns against TB.5−7 In this letter, we describe our efforts to explore the structure–activity relationship (SAR) and structure–property relationship (SPR) of this class and the results of in vivo pharmacokinetics and pharmacological evaluation of selected compounds in mice.
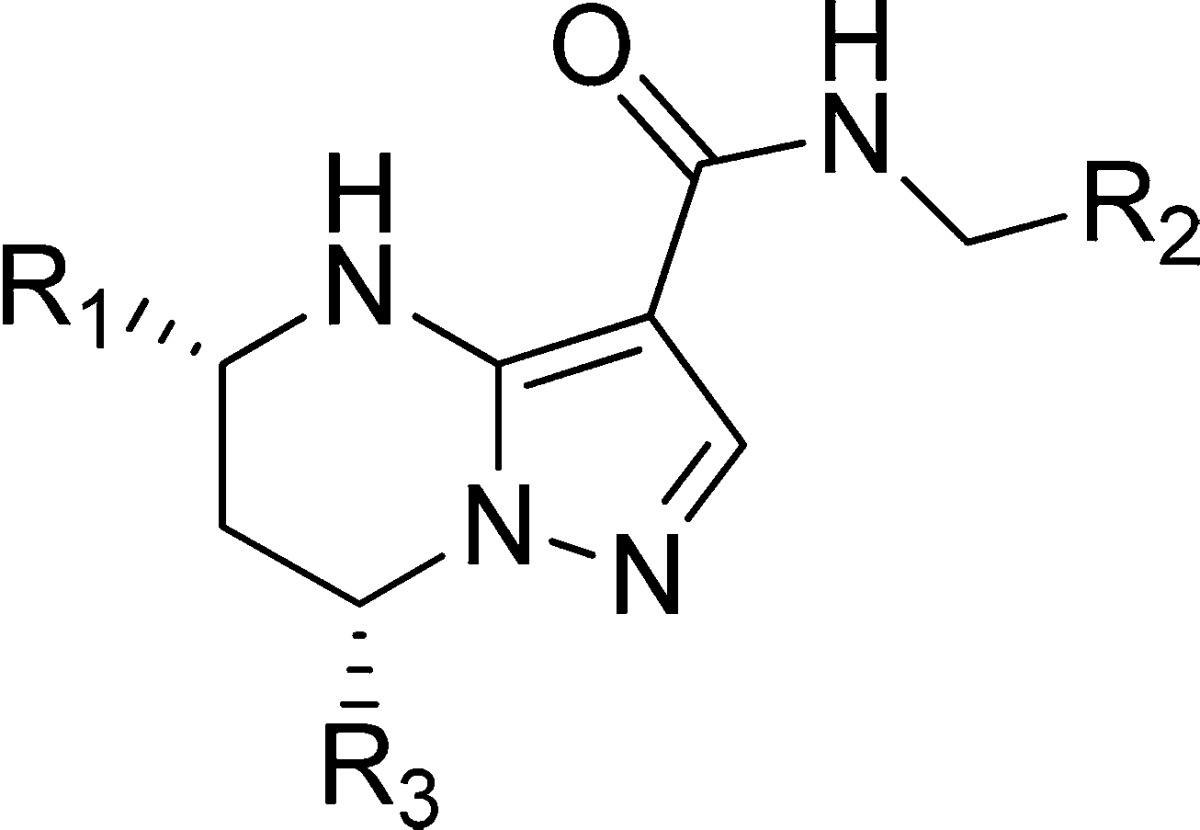
Our initial SAR study identified the key pharmacophore required for anti-TB activity as summarized in Figure 1. The NH of the tetrahydropyrimidine ring, the secondary amide linker, and the pyrazole ring were all found to be essential to retain low micromolar values of MIC (minimal inhibitory concentration, defined as the concentration that prevents 50% of bacterial growth at 5 days postinhibitor exposure). Significant differential anti-TB activity of the stereoisomers at the C-5 and C-7 positions was observed, and the absolute stereochemistry of the active enantiomer was confirmed to be 5R,7S with X-ray crystal analysis (X-ray data can be found in the Supporting Information). The corresponding (5S,7R) isomers were proved to be inactive (MIC > 20 μM). This result indicates that the (5R,7S) form of this scaffold may interact appropriately with some pocket of the as yet unknown biological target inside the TB bacteria. Next, we focused on exploration of SAR and SPR for the phenyl left-hand side (LHS), benzyl right-hand side (RHS), and trifluoromethyl at the C-7 position to optimize the balance of potency and physicochemical properties.
Figure 1.

Initial SAR for tetrahydropyrazolopyrimidines.
The general synthesis of tetrahydropyrazolopyrimidine carboxamide derivatives is shown in Scheme 1.8−11 Condensation of the aminopyrazole 2 with the corresponding diketones 1 in acetic acid yielded the pyrazolopyrimidines 3 as single regioisomer at the 5,7-position. Reduction of the pyrimidine ring with sodium borohydride (NaBH4) afforded only the 5,7-cis isomer of the tetrahydropyridimine analogue 4. The para-methoxy group of 3e was cleaved by boron tribromide (BBr3) to give the phenol 4e. Subsequently, alkaline hydrolysis of the ester 4 with potassium hydroxide afforded the racemic acid 5, which was separated by preparative high-performance liquid chromatography (HPLC) using a chiral column to provide the desired (5R,7S) form 6. Coupling with the corresponding benzyl amines using 2-(1H-7-azabenzotriazole-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU) as a coupling reagent produced the target compounds. The synthesis of compounds 12, 13, 14, and 19 is described in Scheme 2. Introduction of morpholine at the para-position of LHS phenyl was achieved by palladium catalyzed amination of para-bromophenyl of LHS 8(12) to afford compound 12. Compounds 13, 14, and 19 were prepared by alkylation of the para-phenol of LHS 7 with the appropriate alkylating agents.
Scheme 1. General Scheme for Synthesis of Tetrahydropyrazolopyrimidines.

Reagents and conditions: (a) AcOH at 110 °C; (b) NaBH4, EtOH at room temperature; (c) KOH, aq. EtOH at 60 °C; (d) preparative chiral HPLC; (e) HATU, i-PrNEt2, DMF.
Scheme 2. Synthesis of Tetrahydropyrazolopyrimidines.

Reagents and conditions: (a) (i) NaBH4, EtOH at room temperature, (ii) BBr3, CHCl3 at room temperature; (b) KOH, aq. EtOH at 60 °C; (c) preparative chiral HPLC; (d) HATU, i-PrNEt2, DMF; (e) 2-methoxy-bromoethane, Cs2CO3, DMF (for 13 and 19), oxetan-3-yl 4-methylbenzenesulfonate, K2CO3, DMF (for 14); (f) morpholine, Pd(OAc)2, Xantphos, Cs2CO3, toluene (for 12).
Compound 9 exhibited best potency against Mtb H37Rv in the whole cell assay (MIC 0.15 μM); however, it is highly lipophilic (log P = 6.3)13 and shows high plasma protein binding (>99.0%) and low aqueous solubility (<4 μM at pH 6.8), which are in general unfavorable drug-like properties. To reduce the lipophilicity of the scaffold, replacement of the LHS-phenyl with 2-pyridyl and 2-furyl groups led to compounds 10 and 11, which were tolerated and reduced log P significantly (by 0.8–1.9). Introduction of polar substituents at the para-position of the LHS phenyl afforded compounds 12, 13, and 14, which also reduced log P and achieved anti-TB activity comparable with compound 9. Introduction of the 2- and 3-pyridyl rings on the RHS reduced log P without affecting the potency (compounds 15 and 16). However, all of these modifications had little effect on solubility and plasma protein binding. Replacement of the core 7-trifluoromethyl substituent with difluoromethyl in 10 afforded compound 17, which interestingly also increased aqueous solubility. The combination of the LHS pyridyl with RHS pyridyl generated compound 18, which led to a significant decrease in log P (3.3) and thereby increased intrinsic aqueous solubility (0.21 g/L). However, this modification also resulted in the loss of anti-TB activity (MIC = 52.2 μM). Compound 19 suffered from modest anti-TB activity despite its improved physicochemical properties (Table 1).
Table 1. SAR and SPR of the Tetrahydropyrazolopyrimidines Analogues.

Log P: High-throughput measured octanol/water partition coefficient.
Solubility: high throughput equilibrium solubility.
PPB: plasma protein binding measured by rapid equilibrium dialysis (RED) device.
Compound 9 showed a potent bacteriocidal effect and activity in an in vitro macrophage model (further details will be reported in due course). Furthermore, 9 is active across all MDR TB isolates14 suggesting a novel mechanism of action. Studies to elucidate a mechanism of action of this series will be discussed elsewhere.14
The in vivo pharmacokinetics (PK) of compounds 9, 10, 14, and 17 were evaluated in mice by oral (po) and intravenous (iv) routes at doses of 25 and 5 mg/kg, respectively (Table 2). All four compounds displayed low to moderate total systemic clearance and volume of distribution with elimination half-lives ranging from 1.3 to 4 h. These compounds showed good oral bioavailability (45–100%) and good oral exposure in systemic circulation. In addition, these compounds exhibited no significant CYP inhibition (based on reversible inhibition assays using midazolam for CYP 3A4/5, bufuralol for CYP 2D6, and diclofenac for CYP 2C9 as markers) and induction (based on pregnane X-receptor induction assay) in vitro, indicating a low potential risk for in vivo drug–drug interactions. Encouraged by the high potency and promising in vivo PK profile of these compounds, we conducted in vivo efficacy studies in an acute mouse model of Mtb infection.4 Seven days post Mtb infection, the compounds were orally administrated at 100 mg/kg once daily for 28 days. Compound 9 produced a 3.5 log CFU reduction as compared with the untreated control, although surprisingly the related compounds (10, 14, and 17) with similar in vitro activity did not demonstrate any efficacy in vivo. Although we have not yet clearly understood the reason why the rest of the compounds were not efficacious, their shorter half-lives, lower volume distributions, and/or lower lung to plasma distribution (Table 2) may have potential negative effect on efficient delivery of the compounds to the primary site Mtb infection in lungs. It is noteworthy that 9 exhibited comparable oral efficacy with ethambutol at a dose of 100 mg/kg (Figure 2).
Table 2. In Vitro and in Vivo PK Evaluation of the Tetrahydropyrazolopyrimidines Analogues.
| iv PK parametersa |
po PK parametersa |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compounds | in vitro CLd (μL/min/mg) | in vivo CL (mL/min/kg) | Vss (L/kg) | t1/2 (h) | AUC (μM·h) | Cmax (μM) | F (%) | AUC (L/P ratio) | Cmax (L/P ratio) |
| 9 | 43.5 | 31 | 6.3 | 4.2 | 18.9 (91.1)c | 3.6 (8.3)c | 65 | 4.5 | 3.2 |
| 10 | 71.82 | 38 | 3.4 | 1.3 | 25.0 (91.9)b | 3.6 (11.8)b | 100 | 2.1 | 1.9 |
| 14 | 8.92 | 9.9 | 2.5 | 4.1 | 41.7 (71.3)b | 3.7 (7.3)b | 52 | 2.2 | 2.7 |
| 17 | 85.56 | 24 | 2.6 | 1.8 | 16.8 (77.7)b | 5.5 (15.2)b | 53 | 2.8 | 1.7 |
Dose: 5 mg/kg, iv, 25 mg/kg, po; the value in parentheses is at a dose of 100.
The value in parentheses is at a dose of 200.
In mg/kg. L/P ratio: total lung to plasma ratio.
Metabolic stability in mouse liver microsomes.
Figure 2.

In vivo efficacy results in TB mouse model.
In conclusion, we have identified the tetrahydropyrazolopyrimidine scaffold from an Mtb whole cell HTS campaign and examined SAR and SPR of this scaffold. This class has shown good DMPK properties in mouse and one of the compounds demonstrated potent oral efficacy in an acute TB mouse model and has no significant CYP inhibition or induction. All of the data suggest that we have identified a promising candidate, 9, and it has a potential for inclusion in combination therapies for both drug-sensitive and drug-resistant TB.
Acknowledgments
We would like to thank Carolyn Shoen and Michael Cynamon for in vivo mouse efficacy studies and other NITD colleagues for their support during the course of this study.
Supporting Information Available
Full experimental details for compounds synthesized, descriptions of assays, PK data, and in vivo pharmacology results. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ California Institute for Biomedical Research (Calibr), 11119 North Torrey Pines Road, Suite 100, La Jolla, California 92037, United States.
Author Present Address
⊥ Institut Pasteur Korea 696 Sampyeong-dong, Bundang-gu, Seongnam-si, Gyeonggi-do 463-400, Korea.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization. Multidrug and Extensively Drug-Resistant TB: 2010 Global Report on Surveillance and Response.
- Ma Z.; Lienhardt C.; McIlleron H.; Nunn A. J.; Wang X. Global tuberculosis drug development pipeline: the need and the reality. Lancet 2010, 375, 2100–2109. [DOI] [PubMed] [Google Scholar]
- Koul A.; Arnoult E.; Lounis N.; Guillemont J.; Andries K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [DOI] [PubMed] [Google Scholar]
- Pethe K.; Sequeira P. C.; Agarwalla S.; Rhee K.; Kuhen K.; Phong W. Y.; Patel V.; Beer D.; Walker J. R.; Duraiswamy J.; Jiricek J.; Keller T. H.; Chatterjee A.; Tan M. P.; Ujjini M.; Rao S. P.; Camacho L.; Bifani P.; Mak P. A.; Ma I.; Barnes S. W.; Chen Z.; Plouffe D.; Thayalan P.; Ng S. H.; Au M.; Lee B. H.; Tan B. H.; Ravindran S.; Nanjundappa M.; Lin X.; Goh A.; Lakshminarayana S. B.; Shoen C.; Cynamon M.; Kreiswirth B.; Dartois V.; Peters E. C.; Glynne R.; Brenner S.; Dick T. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat. Commun. 2010, 1, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddry J. A.; Ananthan S.; Goldman R. C.; Hobrath J. V.; Kwong C. D.; Maddox C.; Rasmussen L.; Reynolds R. C.; Secrist J. A. III; Sosa M. I.; White E. L.; Zhang W. Antituberculosis activity of the molecular libraries screening center network library. Tuberculosis 2009, 89, 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballel L.; Bates R. H.; Young R. J.; Alvarez-Gomez D.; Alvarez-Ruiz E.; Barroso V.; Blanco D.; Crespo B.; Escribano J.; Gonzalez R.; Lozano S.; Huss S.; Santos-Villarejo A.; Martin-Plaza J. J.; Mendoza A.; Rebollo-Lopez M. J.; Remuinan-Blanco M.; Lavandera J. L.; Perez-Herran E.; Gamo-Benito F. J.; Garcia-Bustos J. F.; Barros D.; Castro J. P.; Cammack N. Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem 2013, 8, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ruiz E.; Ballell-Pages L.; Castro-Pichel J.; Encinas L.; Esquivias J.; Gamo-Benito F. J.; Garcia-Palancar M. C.; Remuinan-Blanco M. J.. Tetrahydropyrazolo [1,5-a] Pyrimidine As Anti-Tuberculosis Compounds. Patent WO2012/143522.
- Dalinger I. L.; Vatsadse I. A.; Shevelev S. A.; Ivachtchenko A. V. Liquid-phase synthesis of combinatorial libraries based on 7-trifluoromethyl-substituted pyrazolo [1,5-a] pyrimidine scaffold. J. Comb. Chem. 2005, 7, 236–245. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Mori A.; Inaba A.; Oka M.; Makino H.; Yamaguchi M.; Fujita H.; Kawamoto T.; Goto M.; Kimura H.; Baba A.; Yasuma T. Synthesis and structure–activity relationship of tetrahydropyrazolopyrimidine derivatives: A novel structural class of potent calcium-sensing receptor antagonists. Bioorg. Med. Chem. 2010, 18, 8501–8511. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Mori A.; Kotani E.; Oka M.; Makino H.; Fujita H.; Ban J.; Ikeda Y.; Kawamoto T.; Goto M.; Kimura H.; Baba A.; Yasuma T. Discovery of novel and potent orally active calcium-sensing receptor antagonists that stimulate pulselike parathyroid hormone secretion: Synthesis and structure–activity relationships of tetrahydropyrazolopyrimidine derivatives. J. Med. Chem. 2011, 54, 1430–1440. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Mori A.; Morimoto S.; Kotani E.; Oka M.; Notoya K.; Makino H.; Ono M.; Shirasaki M.; Tada N.; Fujita H.; Ban J.; Ikeda Y.; Kawamoto T.; Goto M.; Kimura H.; Baba A.; Yasuma T. Novel and potent calcium-sensing receptor antagonists: Discovery of (5R)-N-[1-ethyl-1-(4-ethylphenyl)propyl]-2,7,7-trimethyl-5-phenyl-4,5,6,7-tetrahydropyrazolo [1,5-a] pyrimidine-3-carboxamide monotosylate (TAK-075) as an orally active bone anabolic agent. Bioorg. Med. Chem. 2011, 19, 1881–1894. [DOI] [PubMed] [Google Scholar]
- Klingensmith L. M.; Strieter E. R.; Barder T. E.; Buchwald S. L. New insights into Xantphos/Pd-Catalyzed C–N bond forming reactions: A structural and kinetic study. Organometallics 2006, 25, 82–91. [Google Scholar]
- Measured value. The calculated log P of this scaffold (clog P of compound 9 is 3.3) is significantly lower than the measured log P probably due to the intramolecular hydrogen bond interaction of the NH at the 4-position with the amide carbonyl group.
- Bifani P.; Lakshminarayana S.; Rao S.; Ma I.; Tan B. H.; Yokokaya F.; Kuhen K.; Thayalan P.; Chua A.; Ng S. H.; Cyrille K.; Wong J.; Sherman D.; Pethe K.; Glynne R.; Beer D.; Walker J. R.; Smith P.; Jiricek, J.. High throughput identification of a novel lethal anti-tubercular Tetrahydropyrazolopyrimidine Carboxamide and its mechanisms of inhibition and resistance. European Molecular Biology Organization (EMBO), manuscript in preparation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.