

Abstract

The synthesis and preclinical characterization of two novel, brain penetrating P2X7 compounds will be described. Both compounds are shown to be high potency P2X7 antagonists in human, rat, and mouse cell lines and both were shown to have high brain concentrations and robust receptor occupancy in rat. Compound 7 is of particular interest as a probe compound for the preclinical assessment of P2X7 blockade in animal models of neuro-inflammation.
Keywords: P2X7, neuro-inflammation, depression
The P2X7 ion channel is a member of a large purinoreceptor family that includes both P2X ionotropic and P2Y metabotropic receptors.1 There are seven known P2X receptor subtypes, and of that group, P2X7 has been shown to be involved in the release pro-inflammatory cytokines, including IL-1β.2 As such, numerous reports have appeared in the literature describing the role of P2X7 in a variety of pro-inflammatory disease states including pain, osteoarthritis, rheumatoid arthritis, and pathology associated with neuro-inflammation such as in epilepsy, multiple sclerosis, and a variety of neuro-degenerative states including Alzheimer’s disease.3 Given that the P2X7 receptor is expressed in the CNS on astrocytes and microglial cells and that the expression and activation of P2X7 in glial cells may regulate glutamate and IL-1β release, our interest in this target has focused on the role of P2X7 in neuro-immune modulation. To that end, it has been shown by various groups that the pro-inflammatory cytokine IL-1β is involved in chronic stress models of affective disorders.4−7 Furthermore, P2X7 antagonism has been reported to be efficacious in animal models of mood disorders and mouse/human genetics study link P2X7 locus to mood disorders.8,9 We were particularly interested in recent reports showing evidence that IL-1 receptor blockade may be an effective approach for the treatment of depression, suggesting that a brain-penetrating P2X7 antagonist that blocked IL-1β release in glial cells might be efficacious in mood disorders.10
A wide variety of P2X7 antagonists have been disclosed over the past several years11 including numerous interesting benzamides. Some early examples of drug-like P2X7 antagonists disclosed by the Astra-Zeneca group are the adamantyl-based compounds 1(12) and 2(13) shown below. Both are potent P2X7 antagonists as demonstrated by inhibition of BzATP induced IL1-β release in human peripheral blood monocytes (pA2 = 8.8 and 8.0).

Pfizer also reported on a series of 2-chlorobenzamides discovered via high throughput screening. Compound 3 was reported to be a relatively weak screening hit (P2X7 Yo-Pro IC50 = 1.1 μM); however, medicinal chemistry efforts lead to the identification of an analogue (CE-224,535), a very potent P2X7 antagonist that became a clinical candidate.14 Pfizer recently reported clinical results with CE-224,535 in rheumatoid arthritis patients inadequately controlled by methotrexate.15

While the compound did not show efficacy in a three month rheumatoid arthritis trial, it is of note that Pfizer reported that the compound exposure exceeded the amount required for sustained inhibition of P2X7 (as measured by IL-1β inhibition) for the entire three month period. This data indicates that P2X7 inhibition is unlikely to be a viable approach to the treatment of rheumatoid arthritis; however, it also indicates that sustained inhibition of P2X7 in humans is not likely to be plagued with mechanism-based adverse events.
Examples of other interesting small, drug-like P2X7 antagonists include the imidazolidinecarboxamides and pyroglutamates recently reported by Glaxo SmithKline.16,17 While these compounds are potent human P2X7 antagonists, they also have some activity at the rat P2X7 receptor and therefore could be used for preclinical efficacy studies. The authors report that brain penetrating P2X7 antagonists such as those shown below are efficacious in preclinical pain models.

Our interest in P2X7 antagonists focused on the identification of potent, brain penetrating compounds with high affinity for both the rat and human P2X7 in order to assess their utility for the treatment of CNS disorders, including depression. Toward this end, we now report the discovery of N-((4-(4-phenyl-piperazin-1-yl)tetrahydro-2H-pyran-4-yl)methyl)-2-(phenyl-thio) nicotinamide (7) and 2-methyl-N-((1-(4-phenylpiperazin-1-yl)cyclohexyl)methyl)-1,2,3,4-tetrahydroisoquinoline-5-carbox-amide (8). These compounds were identified after extensive medicinal chemistry efforts following an HTS campaign that showed that simple N-(cyclohexylmethyl)benzamides were weak P2X7 antagonists. The compounds were synthesized as shown in Schemes 1 and 2. For compound 7, 2-(phenylthio)nicotinic acid was first converted to the acid chloride 9. Next, dihydro-2H-pyran-4(3H)-one was condensed with 1-phenylpiperazine and potassium cyanide then reduced to form the amine 10. Condensing 9 and10 then provided compound 7 in reasonable overall yield.
Scheme 1. Synthesis of Compound 7.

Reagents and conditions: (a) oxalyl chloride, dichloromethane, dimethylformamide, 100%; (b) KCN, H2O, pH = 3; (c) lithium aluminum hydride, tetrahydrofuran, 75%; (d) dichloromethane, 53%.
Scheme 2. Synthesis of Compound 8.

Reagents and conditions: (a) methanol, H2O, NaOH, 100%; (b) KCN, H2O, pH = 3; (c) lithium aluminum hydride, tetrahydrofuran, 75%; (d) BOP, triethyl amine, dichloromethane, 77%; (e) trifluoroacetic acid; (f) formaldehyde, sodium triacetoxyborohydride, 74%.
Similarly, compound 8 was prepared from 2-tert-butyl 5-methyl 3,4-dihydroisoquinoline-2,5(1H)-dicarboxylate and the amine 12, which was formed by reaction of cyclohexanone with 1-phenylpiperazine as shown in Scheme 2.
In vitro data for compounds 7 and 8 are detailed in Table 1. Both compounds are potent P2X7 antagonists in human and rodent cell lines, and both compounds show inhibition of Bz-ATP induced IL-1β secretion in human peripheral blood monocytes and in human whole blood. Because both compounds have good functional activity and binding affinity in rat, we chose to further characterize these compounds in vivo with the intention of using one or both of these as tool compounds for pharmacodynamic studies.
Table 1. In Vitro Pharmacology for Compounds 7 and 8.
| human FLIPR pIC50a | rat FLIPR pIC50b | mouse FLIPR pIC50c | human pKi | rat pKi | rat/human SERTd pKi (nM) | human PBMC pIC50e | human WB pIC50f | |
|---|---|---|---|---|---|---|---|---|
| 7 | 8.3 ± 0.08 | 7.2 ± 0.08 | 7.5 ± 0.1 | 7.9 ± 0.08 | 8.7 ± 0.08 | 5.5/n.d. | 7.6 ± 0.07 | 6.7 ± 0.07 |
| 8 | 7.7 ± 0.07 | 7.8 ± 0.1 | 7.1 ± 0.2 | 7.9 ± 0.08 | 9.1 ± 0.07 | 7.7 (n = 2)/7.8 | 7.4 ± 0.07 | 6.7 ± 0.09 |
Human FLIPR pIC50 measured in a Ca2+ flux assay.
Rat FLIPR pIC50 measured in a Ca2+ flux assay.
Mouse FLIPR pIC50 measured in a Ca2+ flux assay.
Rat and human SERT Ki.
Human peripheral blood monocyte pIC50 for IL-1b inhibition.
Human whole blood pIC50, all data are the result of at least three assays run in triplicate ± SEM.
During the course of this characterization, we discovered that compound 8 is a high affinity SERT inhibitor, both in human and rat (Table 1). Both compounds were also screened in a commercial panel of 50 receptor, ion channel, and transporter assays (CEREP, www.cerep.com) at 1 μM. Compound 7 had a <50% effect at all targets tested, whereas compound 8 had >50% inhibition on human NK2 (68% inhib.), human DAT (93% inhib.), and r NaCH (54% inhib.) IC50s were generated for DAT (290 nM) and r NaCH (910 nM). Recognizing that the SERT affinity observed with 8 may be an issue when attempting to assess the antidepressant effects of our P2X7 inhibitors, most of our subsequent efforts were focused on compound 7; however, we continued to profile both lead molecules in order to assess their ability to distribute into the brain.
Prior to any in vivo work, we obtained preliminary DMPK and developability measurements for each compound. The data are shown in Table 2. Both compounds are relatively drug-like with reasonable physical properties, solubility, protein binding, and permeability; however, both suffer from very high extraction ratios in human and rat liver microsomes indicating that they are very unlikely to be suitable for oral delivery. Because subcutaneous dosing is preferred for many in vivo models, we decided to assess the plasma and brain exposures following subcutaneous dosing.
Table 2. Physical Properties and in Vitro DMPK Parameters for Compounds 7 and 8.
| MW | c log P | human ERa | rat ERa | mouse ERa | human/rat ppbb | Caco-2 A to B/B to Ac | brain pbd | Solubility pH2/pH7e | |
|---|---|---|---|---|---|---|---|---|---|
| 7 | 488.7 | 4.3 | >0.95 | >0.92 | >0.93 | 97.8/97.1 | 98.3 | 400/31 | |
| 8 | 446.6 | 5.1 | 0.90 | 0.86 | 0.90 | 83.5/95.6 | 11/2.1 | 97.6 | 95/90 |
Human, rat, or mouse extraction ratio as measured in a microsomal preparation.
Human or rat protein binding reported as % bound.
Papp reported in units of cm/sec × 10–6 (data generated at CEREP, www.cerep.com).
Rat brain protein binding reported as % bound.
Reported in μM.
Initial experiments are shown in Figures 1 and 2. In these experiments, the compound was dosed subcutaneously (s.c.) and plasma and brain concentrations were measured over time, along with central P2X7 receptor occupancy as assessed by ex vivo autoradiography. The results for compound 7 are shown in Figure 1. High P2X7 receptor occupancy was achieved following a single dose of 30 mg/kg s.c. and greater than >50% occupancy remained for 2 h at this dose, indicating robust target engagement. Similar results were seen with 10 mg/kg s.c. of compound 8 (Figure 2).
Figure 1.
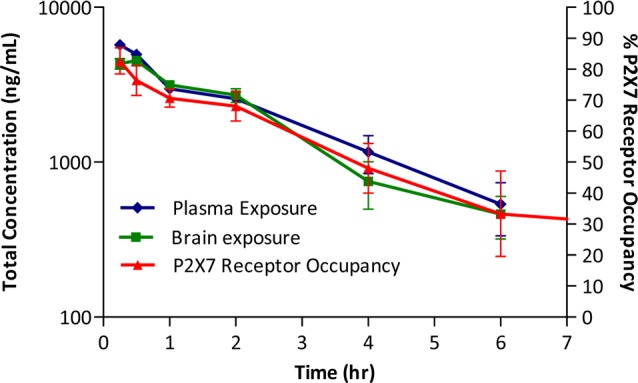
Ex vivo P2X7 receptor occupancy, plasma, and brain levels with compound 7 (30 mg/kg) in rat brain: time dependency after subcutaneous administration (n = 3 per time point ± SEM).
Figure 2.
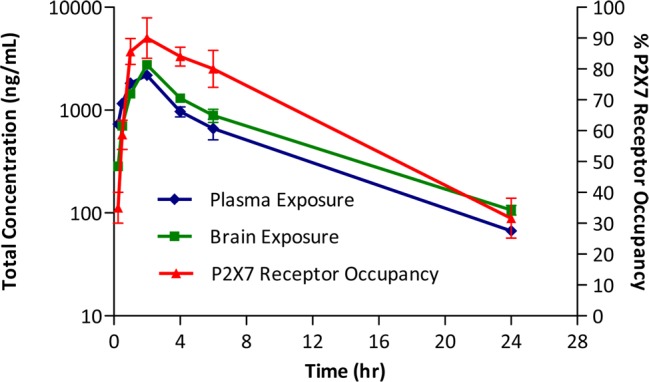
Ex vivo P2X7 receptor occupancy, plasma, and brain levels with compound 8 (10 mg/kg) in rat brain: time dependency after subcutaneous administration (n = 3 per time point ± SEM).
The dose dependency of central receptor occupancy was assessed in a second series of experiments (Figures 3 and 4). Central occupancy was measured at the tmax for each compound (15 min for compound 7 and 120 min for compound 8; data not shown). Compound 7 had an ED50 for occupancy of 2.5 mg/kg, whereas compound 8 had a nearly 10-fold lower ED50 of 0.3 mg/kg. In separate ex vivo autoradiography studies, compound 8 was determined to have ED50 for SERT occupancy of 24 mg/kg, indicating that SERT target engagement in rat was not significant at doses where robust P2X7 receptor occupancy was observed.
Figure 3.

Ex vivo P2X7 receptor occupancy with compound 7 in rat brain: dose dependency following subcutaneous administration (n = 3 per dose ± SEM). P2X7 occupancy was measured 15 min after drug administration.
Figure 4.

Ex vivo P2X7 receptor occupancy with compound 8 in rat brain: dose dependency following subcutaneous administration (n = 3 per dose ± SEM). P2X7 occupancy was measured 120 min after drug administration.
In conclusion, we have demonstrated that compounds 7 and 8 are high affinity rat, mouse, and human P2X7 receptor antagonists and that both compounds have DMPK properties suitable for preclinical pharmacodynamics studies. At appropriate doses, both compounds were shown to occupy central P2X7 receptors in vivo, as assessed by ex vivo autoradiography studies. Future reports on the pharmacology of these interesting P2X7 compounds will focus on the characterization of compound 7 in a variety of preclinical models of depression and related mood disorders.
Glossary
Abbreviations
- IL-1β
interleukin-1β
- Bz-ATP
benzoyl adenosine triphosphate
- SERT
serotonin transporter
- DAT
dopamine transporter
Supporting Information Available
Detailed synthetic procedures for all intermediates and products; descriptions of assays used to characterize compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ralevic V.; Burnstock G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998, 50, 413. [PubMed] [Google Scholar]
- Ledeboer A.; Sloane E. M.; Milligan E. D.; Frank M. G.; Mahony J. H.; Maier S. F.; Watkins L. R. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain 2005, 115, 71–83. [DOI] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Cruz-Lopez O.; Lopez-Cara C.; Preti D.; Borea P. A.; Gessi S. The P2X7 receptor as a therapeutical target. Expert Opin. Ther. Targets 2008, 125647–661. [DOI] [PubMed] [Google Scholar]
- Goshen I.; Kreisel T.; Ben-Menachem-Zidon O.; Licht T.; Weidenfeld J.; Ben-Hur T.; et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol. Psychiatry 2008, 137717–728. [DOI] [PubMed] [Google Scholar]
- Koo J. W.; Duman R. S. Evidence for IL-1 receptor blockade as a therapeutic strategy for the treatment of depression. Curr. Opin. Invest. Drugs 2009, 107664–671. [PMC free article] [PubMed] [Google Scholar]
- Koo J. W.; Duman R. S. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Natl. Acad. Sci. U.S.A. 2008, 1052751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K. A.; Thomsen C. The role of the innate immune system in psychiatric disorders. Mol. Cell Neurosci. 2013, 53, 52–62. [DOI] [PubMed] [Google Scholar]
- Csolle C.; Ando R. D.; Kittel A.; Goloncser F.; Baranyi M.; Soproni K.; Zelena D.; Haller J.; Nemeth T.; Mocsai A.; Sperlagh B. The absence of P2 × 7 receptors (P2rx7) on non-haematopoietic cells leads to selective alteration in mood-related behaviour with dysregulated gene expression and stress reactivity in mice. Int. J. Neuropsychopharmacol. 2013, 161213–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M.; Ota K. T.; Duman R. S. The inflammasome: Pathways linking psychological stress, depression, and systemic illnesses. Brain, Behav., Immun. 2013, 10.1016/j.bbi.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo J. W.; Duman R. S. Evidence for IL-1 receptor blockade as a therapeutic strategy for the treatment of depression. Curr. Opin. Invest. Drugs 2009, !07664–671. [PMC free article] [PubMed] [Google Scholar]
- Guile S. D.; Alcaraz L.; Birkinshaw T. N.; Bowers K. C.; Ebden M. R.; Furber M.; Stocks M. J. Antagonists of the P2X7 Receptor. From Lead Identification to Drug Development. J. Med. Chem. 2009, 52, 3123–3141. [DOI] [PubMed] [Google Scholar]
- Baxter A.; Bent J.; Bowers K.; Braddock M.; Brough S.; Fagura M.; Lawson M.; McInally T.; Mortimore M.; Robertson M.; Weaver R.; Webborn P. Hit-to-lead studies: The discovery of potent adamantane amide P2X7 receptor antagonists. Bioorg. Med. Chem. Lett. 2003, 4047–4050. [DOI] [PubMed] [Google Scholar]
- Furber M.; Alcaraz L.; Bent J. E.; Beyerbach A.; Bowers K.; Braddock M.; Caffrey M. V.; Cladingboel D.; Collington J.; Donald D. K.; Fagura M.; Ince F.; Kinchin E. C.; Laurent C.; Lawson M.; Luker T. J.; Mortimore M. M. P.; Pimm A. D.; Riley R. J.; Roberts N.; Robertson M.; Theaker J.; Thorne P. V.; Weaver R.; Webborn P.; Willis P. Discovery of potent and selective adamantane-based small-molecule P2X7 receptor antagonists/interleukin-1β inhibitors. J. Med. Chem. 2007, 50, 5882–5885. [DOI] [PubMed] [Google Scholar]
- Duplantier A. J.; Dombroski M. A.; Subramanyam C.; Beaulieu A. M.; Chang S.-P.; Gabel C. A.; Jordan C.; Kalgutkar A. S.; Kraus K.; Labasi J. M.; Mussari C.; Perregaux D. G.; Shepard R.; Taylor T. J.; Trevena K. A.; Whitney-Pickett C.; Yoon K. Optimization of the physicochemical and pharmacokinetic attributes in a 6-azauacil series of P2X7 receptor antagonists leading to the discovery of the clinical candidate CE-244,535. Bioorg. Med. Chem. Lett. 2011, 21, 3708–3711. [DOI] [PubMed] [Google Scholar]
- Stock T. C.; Bloom B. J.; Wei N.; Ishaq S.; Park W.; Wang X.; Gupta P.; Mebus C. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J. Rheumatol. 2012, 39, 720–727. [DOI] [PubMed] [Google Scholar]
- Abberley L.; Bebius A.; Beswick P. J.; Billinton A.; Collis K. L.; Dean D. K.; Fonria E.; Gleave R. J.; Medhurst S. J.; Michel A. D.; Moses A. P.; Patel S.; Roman S. A.; Scoccitti T.; Smith B.; Steadman J. G. A.; Walter D. S. Identification of 2-oxo-N-(phenylmethyl)-4-imidazolidinecarboxamide antagonists of the P2X7 receptor. Bioorg. Med. Chem. Lett. 2010, 20, 6370–6374. [DOI] [PubMed] [Google Scholar]
- Abdi M. H.; Beswick P. J.; Billinton A.; Chambers L. J.; Charlton A.; Collins S. D.; Collis K. L.; Dean D. K.; Fonfria E.; Bleave R. J.; Lejeune C. L.; Livermore D. G.; Medhurst S. J.; Michel A. D.; Moses A. P.; Page L.; Petal S.; Roman S. A.; Senger S.; Slingby B.; Steadman J. G. A.; Stevens A. J.; Walter D. S. Discovery and structure–activity relationships of a series of pyroglutamic acid amide antagonists of the P2X7 receptor. Bioorg. Med. Chem. Lett. 2010, 20175080–5084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.