

Abstract

Deregulation of HER family signaling promotes proliferation and tumor cell survival and has been described in many human cancers. Simultaneous, equipotent inhibition of EGFR-, HER2-, and HER3-mediated signaling may be of clinical utility in cancer settings where the selective EGFR or HER2 therapeutic agents are ineffective or only modestly active. We describe the discovery of AZD8931 (2), an equipotent, reversible inhibitor of EGFR-, HER2-, and HER3-mediated signaling and the structure–activity relationships within this series. Docking studies based on a model of the HER2 kinase domain helped rationalize the increased HER2 activity seen with the methyl acetamide side chain present in AZD8931. AZD8931 exhibited good pharmacokinetics in preclinical species and showed superior activity in the LoVo tumor growth efficacy model compared to close analogues. AZD8931 is currently being evaluated in human clinical trials for the treatment of cancer.
Keywords: AZD8931, HER receptor family, kinase inhibitor
The HER receptor family comprises four related receptor tyrosine kinases (EGFR, HER2, HER3, and HER4) and is associated with two main ligand classes:1 the first class binds to EGFR, and the second class, which includes heregulins, binds to HER3 and HER4. HER2 lacks ligand-binding capacity. Homo- and/or heterodimerization of HER receptors results in the phosphorylation of key tyrosine residues in the intracellular domain and leads to the stimulation of numerous intracellular signal transduction pathways involved in cell proliferation and survival.2,2b Deregulation of HER family signaling promotes proliferation, invasion, metastasis, angiogenesis, and tumor cell survival and has been described in many human cancers, including those of the lung, head and neck, and breast.3,3b
Therefore, the HER receptor family represents a class of rational targets for anticancer drug development, and a number of small molecules targeting EGFR and HER2 are now clinically available, including gefitinib, erlotinib, and lapatinib (Figure 1). More recently, the importance of the composition of functional HER dimeric units in tumor cell signaling has become apparent in diverse systems, modeling both ligand-dependent and independent drives. Careful profiling of all four HER receptors has differentiated their molecular function,4 and HER3 has been found to have a central role in the transduction of signals to the phosphatidylinositol 3-kinase (PI3K) pathway, thus mediating cell survival signals for EGFR, HER2, and potentially HER4.5 We hypothesized that simultaneous, equipotent inhibition of EGFR-, HER2-, and HER3-mediated signaling may be of clinical utility in cancer settings where the current HER therapeutic agents are ineffective or only modestly active.
Figure 1.

Structure of gefitinib, erlotinib, and lapatinib.
Previous projects at AstraZeneca looking for selective EGFR6−6d or HER27−7d inhibitors led to several preclinical and/or clinical candidates, including gefitinib,6 a selective EGFR kinase inhibitor. Screening our collection of EGFR kinase inhibitors for HER2 activity identified a number of potential lead compounds with HER2 and EGFR inhibitory activity. In this letter, we describe the optimization of one of these leads, compound 1, now also reported in ref (6c), which led to the discovery of AZD8931, an equipotent, reversible inhibitor of signaling by EGFR, HER2, and HER3.
The compounds listed in Tables 1–3 were synthesized from 6-acetoxy 4-chloro 7-methoxyquinazoline6 (see Supporting Information for synthetic schemes, procedures, and characterization of compounds 1–15) and were evaluated in an isolated HER2 kinase assay and/or a ligand-independent HER2 phosphorylation assay in MCF7 cl.24 cells.8
Table 1. Enzyme and Cell Data for Compounds 1–6.
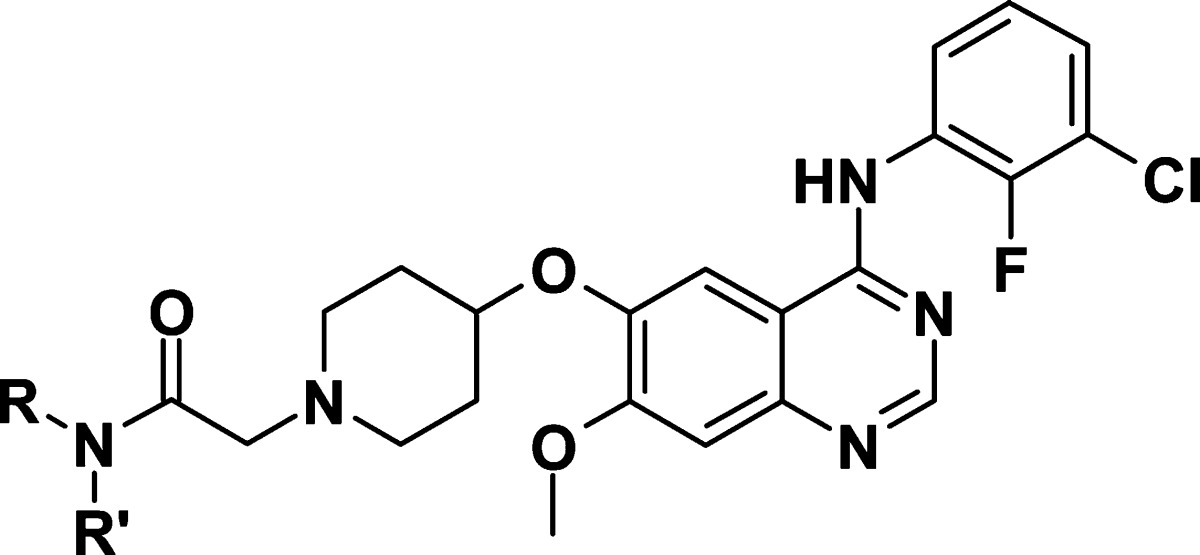
| NRR′ | HER2 enz IC50a | cl.24 IC50 (p-HER2)a | |
|---|---|---|---|
| 1 | NH2 | 0.046 | 0.107 |
| 2 | NHMe | 0.014 | 0.060 |
| 3 | NHEt | 0.033 | 0.12 |
| 4 | NHCH2CH2OMe | 0.091 | 0.25 |
| 5 | NHiPr | 0.11 | 0.41 |
| 6 | NMe2 | 0.22 | 0.67 |
μM; n = 2 or more; CIR values available in the Supporting Information.
Table 3. Cell Data for Compounds 11–15.
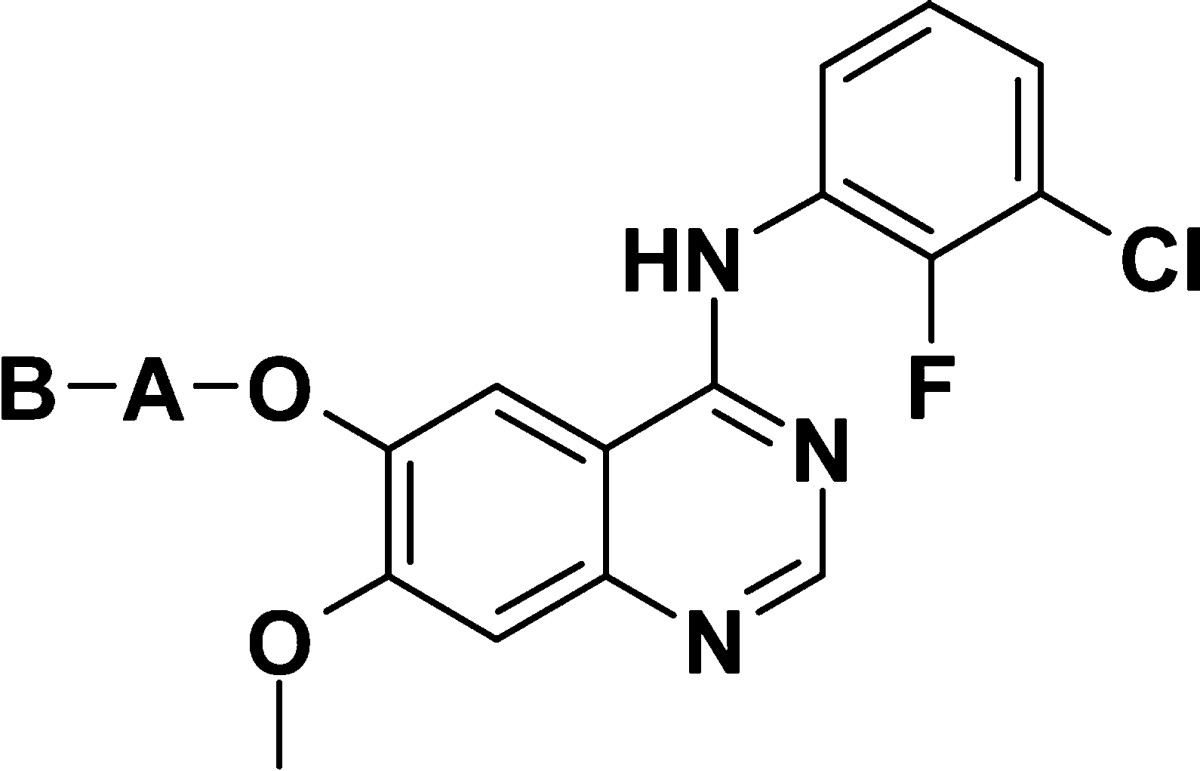
| A | B | cl.24 IC50 (p-HER2)a | |
|---|---|---|---|
| 11 | 4-piperidinyl | CH2CH2CONHMe | 0.550 |
| 12b | 3-piperidinyl | CH2CONHMe | 0.23 |
| 13b | 3-pyrrolidinyl | CH2CONHMe | 0.25 |
| 14 | 3-azetidinyl | CH2CONHMe | 1.1 |
| 15 | –CH2CH2N(Me)– | CH2CONHMe | 1.5 |
μM; n = 2 or more; CIR values available in the Supporting Information.
Evaluated as racemates.
Compound 1 showed potent inhibition of HER2 in both the enzyme and cellular assays (see Table 1) as well as potent inhibition of EGFR (inhibition of KB cell proliferation; IC50 4 nM). Because of its favorable physical (e.g., fraction unbound in rat plasma fu 4.4%) and pharmacokinetic properties (clearance Cl, 16 mL/min/kg; bioavailability F, 27%; from an oral dose of 5 mg/kg and an i.v. dose of 2 mg/kg in AP-Wistar rats), this starting point was considered promising if HER2 activity could be improved. It was found that variation of the amide substitution could lead to changes in HER2 activity. Substitution to the methyl amide 2 showed potency increases both at the HER2 enzymatic and cellular level. Of note, potency was reduced when increasing the size of the amide substituent (i.e., compounds 3–5) further or with a dimethyl amide 6 (see Table 1).
The compounds included in Table 2 illustrate the SAR around the aniline. The 2-fluoro-3-chloroaniline 2 showed improved HER2 potency compared to the 3-chloro-4-fluoroaniline (aniline side chain found in gefitinib) 7,9 the 3-chloroaniline 9, or 2-fluoro-5-chloroaniline 10. Interestingly, the 2,4-difluoro-3-chloroaniline 8 also exhibited good HER2 potency. Similar SAR for fluorine regioisomers was observed on EGFR activity.6c
Table 2. Cell Data for Compounds 7–10.
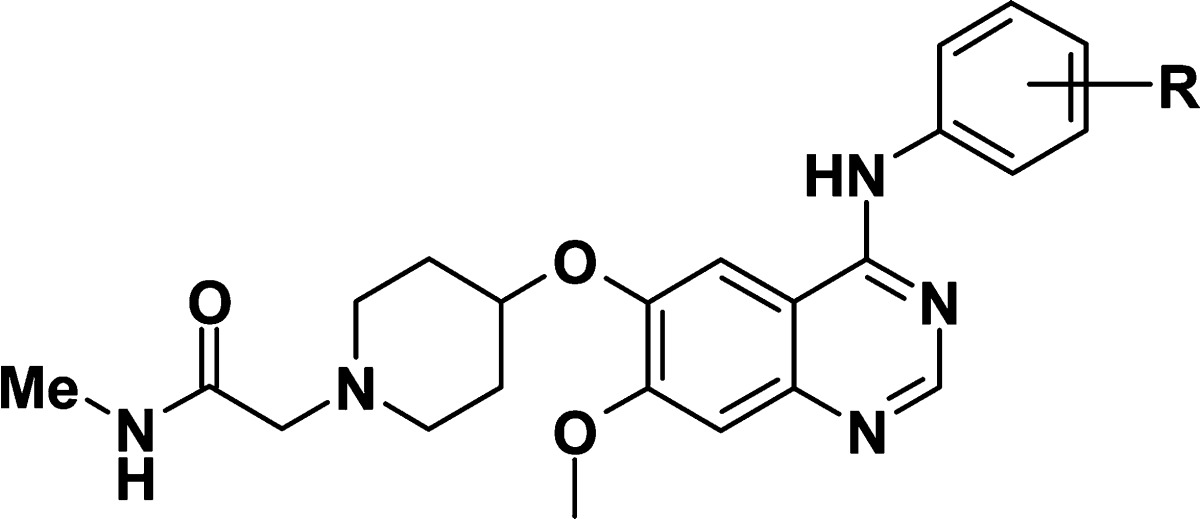
| R | cl.24 IC50 (p-HER2)a | |
|---|---|---|
| 7 | 3-Cl 4-F | 0.264 |
| 8 | 2-F 3-Cl 4-F | 0.089 |
| 9 | 3-Cl | 0.360 |
| 10 | 2-F 5-Cl | 0.860 |
μM, n = 2 or more, CIR values available in the Supporting Information.
As shown in Table 3, modification of the piperidine and the methylene linker at the C-6 position on the quinazoline showed the initial combination of 4-piperidine and a methylene linker to be optimal: addition of a methylene (e.g., 11) or modifications of the cycle (e.g., 12–15) showed reduced activity.
Although the role of the hydrophobic interactions of 4-piperidine in the binding site may also contribute to the increased activity of compound 2 compared to the less lipophilic azetidine 14 or acyclic chain 15, the methyl acetamide side chain appears to be a key requirement for increased HER2 potency. Each modification listed in Table 3 reduces HER2 potency, as it positions the amide in a different region. Disubstitution of (i.e., compound 6) or bulkiness around (i.e., compound 5) the amide reduces HER2 potency. These two observations suggest a key role for the amide NH in binding to the active site.
Most of the reported HER2 inhibitors10 rely on either an extended aniline interacting deep in the selectivity pocket to bring in potency but also compromise physical properties, or covalent binding to a conserved cysteine in the HER kinases, in general via an acrylamide, with potential toxicity risks associated to the reactivity of this functional group. It is the first time that interactions in the solvent channel have been shown to bring in HER2 activity. Therefore, we attempted to rationalize the role of this amide using molecular modeling. Figure 2 shows the anticipated binding mode for compound 2 manually docked into a homology model of HER2 kinase based on the crystal structure of EGFR tyrosine kinase in complex with 4-anilino quinazoline inhibitor erlotinib (PDB code 1M17).11,12 The quinazoline N-1 binds to the hinge region at Met801, with the aniline buried deep in the selectivity pocket. The C6 4-piperidinyloxy sits at the solvent exposed rim of the ATP binding site making various hydrophobic contacts with the surrounding residues (Leu726, Gly727, and Cys805) and short contacts between piperidine CH and backbone carbonyls (Leu726, and Gly727). The piperidine basic site is in the vicinity of the Asp808 residue offering an electrostatic complementarity with the cationic site.
Figure 2.
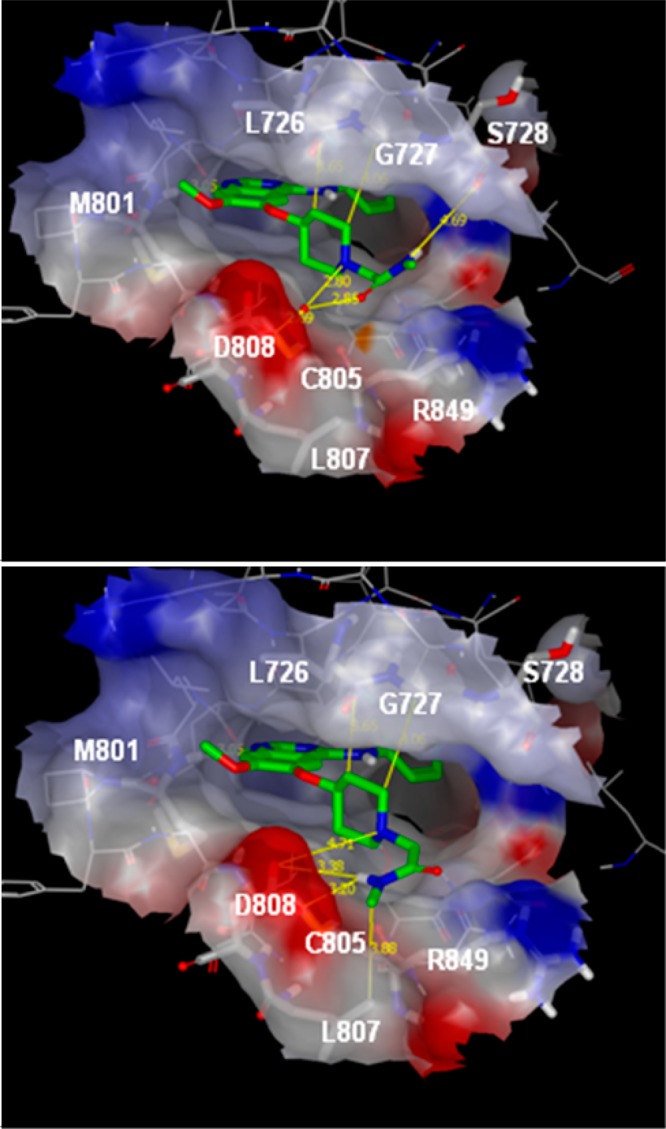
Compound 2 positioned in the HER2 homology model based on the EGFR tyrosine kinase in complex with 4-anilino quinazoline inhibitor erlotinib (PDB code 1M17). The quinazoline N-1 binds to the hinge region at Met801, with the aniline buried deep in the selectivity pocket. The C6 alkoxy 4-piperidine sits at the solvent exposed rim of the ATP binding site making various hydrophobic contacts or CH···C=O short contacts with the surrounding residues (highlighted by yellow lines). The piperidine basic site is in the vicinity of the Asp808 residue offering an electrostatic complementarity with the cationic site. The molecule is shown in two representative conformations of the N-2-acetamide chain, conformation A (upper panel) and conformation B (lower panel). In the model corresponding to conformation A, a water molecule has been added to satisfy the interactions network. The protein surface is colored according to electronic surface potential. Areas in red indicate increasing negative potential, areas in blue increasing positive surface potential.
The precise conformation of the amide NH remains hypothetical. Indeed, depending on the protonation state of the piperidine in the enzyme (piperidine pKa is 6.7 in compound 2), the favored conformation of the acetamide may vary. A search for a piperidine N-2-acetamide scaffold in the small-molecule crystallographic database (CCDC) reveals three main conformations that we have illustrated in Figure 3a.13 In addition to the protonation state, the crystal packing and intermolecular interactions impact on the resulting conformation. On the basis of these results, the three conformations of the piperidine N-2-acetamide substituent of compound 2 were built and docked in our homology model (Figure 2).
Figure 3.

Conformation of piperidine N-2-acetamide scaffold. (a) Three main conformations observed in crystal structures of compounds bearing a piperidine N-2-acetamide chain. (b) Crystal structure of compound 2 as a dihydrate acetonitrile solvate presenting conformation B.
In a type A conformation, the 3D model suggests the amide NH makes an H-bond interaction with the backbone carbonyl of Ser728 in the P-loop, although the H-bond is quite loose. Disubstitution of the amide leads to the loss of H-bond and to a putative steric clash with the loop. However, the amide C=O is oriented toward the Asp808 side chain at a mean distance of 4.5 Å. In the absence of an experimental structure, we cannot exclude the possibility that a water molecule is captured between the intervening heteroatoms and helps to consolidate the interaction network. It is interesting to note that in the closely related HER4 crystal structure in complex with a 4-anilino thieno pyrimidine derivative (PDB code 2R4B) a water molecule is actually present at an analogous location.14d
In a type B conformation, the amide NH points in the direction of the Asp808 carboxylic group making an H-bond interaction. In this orientation, the methyl on the amide makes hydrophobic contact with Leu807, which may offer a rationale for the better potency of compound 2 compared to the primary amide 1 but also explain why bigger substituents than a methyl lead to some loss of potency (i.e., compounds 3, 4, and 5). Moreover, in this conformation, the addition of a second methyl on the amide prevents this type of conformation due to steric clash with the piperidine nitrogen itself.
Interestingly, we successfully obtained a small-molecule X-ray structure of compound 2 as a dihydrate acetonitrile solvate, which presents a type B conformation (Figure 3b). Furthermore, precedents exist for the type B conformation with similar scaffolds in the protein structural database: we identified two kinase structures with a ligand in a type B conformation probably favored by extra interactions with the surrounding residues (PDB code 3CGO and 2B55).14−14c Mindful that the binding mode is based on a modeling approach, the overall conformation B of the N-2-acetamide better fits the observed SAR. Note conformation C has not been discussed here as it does not fit the biological data.
Further biological evaluation8 characterized compound 2 as an equipotent, reversible inhibitor of phosphorylation of EGFR, HER2, and HER3. It showed potent in vitro inhibition against isolated HER2 and EGFR tyrosine kinases (respectively, IC50 14 and 12 nM against HER2 and EGFR). At the cellular level, it inhibited EGF-stimulated phosphorylation of EGFR in the KB cell line (IC50: 4 nM) and heregulin-stimulated phosphorylation of HER2 (IC50: 3 nM) and HER3 (IC50: 4 nM) in the MCF-7 cell line. The broader kinase selectivity of 2 was assessed using diverse panels of serine/threonine, tyrosine, and lipid kinases. This translated into a >1000-fold selectivity for 206 of these enzymes and >100 fold for MNK1 and Flt-3.8 Furthermore, 2 exhibited no CYP P450 inhibition (IC50 > 10 μM against 1A2, 2C9, 2C19, 2D6, and 3A4). It displayed high fraction unbound in rat and human plasma (higher than 1 or 7). The higher fraction unbound seen with the 2-fluoro 3-chloro aniline 2 compared to the 4-fluoro 3-chloro aniline 7 is likely linked to the reduced lipophilicity associated with this aniline (Table 4) and is a pattern observed with a wider set of matched pairs (data not shown).
Table 4. Fraction Unbound (fu) in Rat and Human Plasma and log D7.4 for Compounds 1, 2, and 7.
| fu % (rat) | fu % (human) | log D7.4 | |
|---|---|---|---|
| 1 | 4.4 | 4.7 | 3.1 |
| 2 | 7.2 | 6.7 | 3.2 |
| 7 | 2.9 | 2.7 | 4.0 |
Furthermore, 2 exhibited a pH dependent aqueous solubility due to the two ionizable sites (piperidine pKa 6.7, quinazoline pKa 5.0; aqueous solubility at pH 6.8 (phosphate buffer): 17 μM) and good permeability as measured in CaCo-2 cells (Papp,A to B 15.9 × 10–6 cm·s–1 at 10 μM). Compound 2 displayed favorable oral pharmacokinetics in rat and dog (low clearance and good bioavailability, see Table 5) and low human hepatocyte turnover (Clint < 4.5 μL/min/106 cells). In nude mouse after oral administration at 50 mg/kg, 2 showed improved exposure (AUC: 66 μM·h) compared to 7 and 1 (AUC: 19 μM·h for both).
Table 5. Pharmacokinetic Parameters for 2 in Rat and Dog.
Measured on the free base.
Average of 4 female rats; p.o. 5 mg/kg; i.v. 2 mg/kg.
Average of one male and one female; p.o. 2 mg/kg; i.v. 1 mg/kg.
On the basis of its equipotent pharmacology against EGFR, HER2, and HER3 signaling, its favorable physical properties (high plasma fraction unbound fu, acceptable solubility, and good permeability), and its good pharmacokinetics across preclinical species, 2 was selected for further in vivo evaluation: it showed potent tumor growth inhibition in various xenograft models, driven by EGFR alone (LoVo, PC9) or EGFR and HER2 (BT474C, Calu3, and FaDu) with concomitant pharmacodynamic changes (e.g., phosphorylated EGFR, HER2, and/or HER3).8
Compound 2 was evaluated head to head against 1 and 7 in the LoVo mouse xenograft model: based on high potency against EGFR, better exposure in mouse, and better plasma fraction unbound, it showed better tumor growth inhibition (76% inhibition at 100 mg/kg oral dose once daily vs 36% for 1 and 28% for 7 at the same dose, Figure 4).
Figure 4.
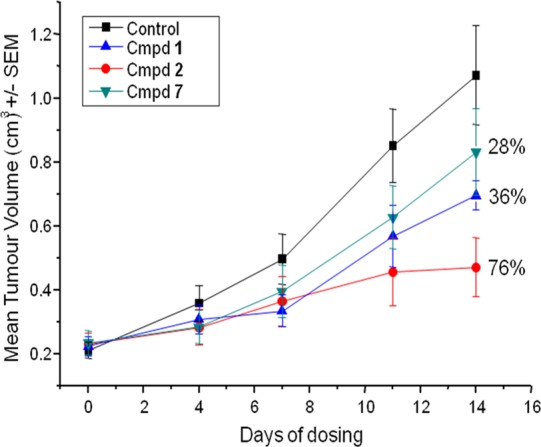
Tumour growth inhibition of 1, 2, and 7 in LoVo human tumor xenografts as measured by mean tumor volume. All three compounds were administered orally once daily using a 100 mg/kg dose. Error bars denote the standard error of the mean.
In conclusion, we report the discovery of 2, an equipotent, reversible inhibitor of signaling by EGFR, HER2, and HER3. Docking studies based on a model of the HER2 kinase domain helped rationalize the increased HER2 activity seen with the methyl acetamide side chain present in compound 2. Compound 2 exhibits good selectivity among kinases outside the HER family and favorable physical properties and pharmacokinetics across species. It exhibits potent tumor growth inhibition in various xenografts models, driven by EGFR alone or by EGFR and HER2. Compound 2 (also known as AZD8931) is currently in phase II clinical trials in patients with solid tumors.
Acknowledgments
We would like to acknowledge the following scientists for their contribution in the synthesis, characterization, and evaluation of the compounds: Annie Olivier, Hervé Germain, Christian Delvare, Delphine Dorison-Duval, Jennifer Vincent, Rowena Callis, Sara Davenport, Helen Ross, Robin Smith, and Frederic Ducrozet. We warmly thank Dr. Ann Ertan (AstraZeneca Södertalje) for determining the X-ray crystal structure of 2.
Supporting Information Available
Details of the synthesis and spectroscopic characterization of all compounds and intermediates together with protocols for biological experiments and crystallographic information. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ The Paterson Institute for Cancer Research, University of Manchester, Wilmslow Road, Manchester M20 4BX, United Kingdom.
Author Present Address
∥ Galderma R&D, les Templiers, 2400 route des Colles, 06410 Sophia-Antipolis, France.
Author Present Address
⊥ Former AstraZeneca employees.
The authors declare no competing financial interest.
Supplementary Material
References
- Olayioye M. A.; Neve R. M.; Lane H. A.; Hynes N. E. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorissen R. N.; Walker F.; Pouliot N.; Garrett T. P.; Ward C. W.; Burgess A. W. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp. Cell. Res. 2003, 284, 31–53. [DOI] [PubMed] [Google Scholar]
- Citri A.; Skaria K. B.; Yarden Y. The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp. Cell. Res. 2003, 284, 54–65. [DOI] [PubMed] [Google Scholar]
- Salomon D.; Gullick W. The erbB family of receptors and their ligands: multiple targets for therapy. Signal 2001, 2, 4–11. [Google Scholar]
- Ciardiello F.; Tortora G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–74. [DOI] [PubMed] [Google Scholar]
- Schulze W. X.; Deng L.; Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol. Syst. Biol. 2005, 1, 2005.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman J. A.; Jänne P. A.; Mermel C.; et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker A. J.; Gibson K. H.; Grundy W.; Godfrey A. A.; Barlow J. J.; Healy M. P.; Woodburn J. R.; Ashton S. E.; Curry B. J.; Scarlett L.; Henthorn L.; Richards L. Studies leading to the identification of ZD1839 (Iressa): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg. Med. Chem. Lett. 2001, 11, 1911–1914. [DOI] [PubMed] [Google Scholar]
- Hennequin L. F. A.; Ballard P.; Boyle F. T.; Delouvrie B.; Ellston R. P. A.; Halsall C. T.; Harris C. S.; Hudson K.; Kendrew J.; Pease J. E.; Ross H. S.; Smith P.; Vincent J. L. Novel 4-anilinoquinazolines with C-6 carbon-linked side chains: Synthesis and structure–activity relationship of a series of potent, orally active, EGF receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 2672–2676. [DOI] [PubMed] [Google Scholar]
- Ballard P.; Bradbury R. H.; Harris C. S.; Hennequin L. F. A.; Hickinson M.; Kettle J. G.; Kendrew J.; Klinowska T.; Ogilvie D. J.; Pearson S. E.; Williams E. J.; Wilson I. Inhibitors of epidermal growth factor receptor tyrosine kinase: Optimization of potency and in vivo pharmacokinetics. Bioorg. Med. Chem. Lett. 2006, 16, 4908–4912. [DOI] [PubMed] [Google Scholar]
- Ballard P.; Bradbury R. H.; Harris C. S.; Hennequin L. F. A.; Hickinson M.; Johnson P. D.; Kettle J. G.; Klinowska T.; Leach A. G.; Morgentin R.; Pass M.; Ogilvie D. J.; Olivier A.; Warin N.; Williams E. J. Inhibitors of epidermal growth factor receptor tyrosine kinase: Novel C-5 substituted anilinoquinazolines designed to target the ribose pocket. Bioorg. Med. Chem. Lett. 2006, 16, 1633–1637. [DOI] [PubMed] [Google Scholar]
- Ballard P.; Bradbury R. H.; Hennequin L. F. A.; Hickinson D. M.; Johnson P. D.; Kettle J. G.; Klinowska T.; Morgentin R.; Ogilvie D. J.; Olivier A. 5-Substituted 4-anilinoquinazolines as potent, selective and orally active inhibitors of erbB2 receptor tyrosine kinase. Bioorg. Med. Chem. Lett. 2005, 15, 4226–4229. [DOI] [PubMed] [Google Scholar]
- Ballard P.; Barlaam B. C.; Bradbury R. H.; Dishington A.; Hennequin L. F. A.; Hickinson D. M.; Hollingsworth I. M.; Kettle J. G.; Klinowska T.; Ogilvie D. J.; Pearson S. E.; Scott J. S.; Suleman A.; Whittaker R.; Williams E. J.; Wood R.; Wright L. Neutral 5-substituted 4-anilinoquinazolines as potent, orally active inhibitors of erbB2 receptor tyrosine kinase. Bioorg. Med. Chem. Lett. 2007, 17, 6326–6329. [DOI] [PubMed] [Google Scholar]
- Barlaam B.; Ballard P.; Bradbury R. H.; Ducray R.; Germain H.; Hickinson D. M.; Hudson K.; Kettle J. G.; Klinowska T.; Magnien F.; Ogilvie D. J.; Olivier A.; Pearson S. E.; Scott J. S.; Suleman A.; Trigwell C. B.; Vautier M.; Whittaker R. D.; Wood R. A new series of neutral 5-substituted 4-anilinoquinazolines as potent, orally active inhibitors of erbB2 receptor tyrosine kinase. Bioorg. Med. Chem. Lett. 2008, 18, 674–678. [DOI] [PubMed] [Google Scholar]
- Barlaam B.; Acton D. G.; Ballard P.; Bradbury R. H.; Cross D.; Ducray R.; Germain H.; Hudson K.; Klinowska T.; Magnien F.; Ogilvie D. J.; Olivier A.; Ross H. S.; Smith R.; Trigwell C. B.; Vautier M.; Wright L. Neutral 5-substituted 4-indazolylaminoquinazolines as potent, orally active inhibitors of erbB2 receptor tyrosine kinase. Bioorg. Med. Chem. Lett. 2008, 18, 1799–1803. [DOI] [PubMed] [Google Scholar]
- Hickinson D. M.; Klinowska T.; Speake G.; Vincent J.; Trigwell C.; Anderton J.; Beck S.; Marshall G.; Davenport S.; Callis R.; Mills E.; Grosios K.; Smith P.; Barlaam B.; Wilkinson R. W.; Ogilvie D. AZD8931, an Equipotent, Reversible Inhibitor of Signaling by Epidermal Growth Factor Receptor, ERBB2 (HER2), and ERBB3: A Unique Agent for Simultaneous ERBB Receptor Blockade in Cancer. Clin. Cancer Res. 2010, 16, 1159–1169. [DOI] [PubMed] [Google Scholar]
- Previously described in Himmelsbach F.; Jung B.; Solca F. Int. Pat. Appl. WO 2003082290.
- Carter C. A.; Kelly R. J.; Giaccone G. Small-molecule inhibitors of the human epidermal receptor family. Expert Opin. Invest. Drugs 2009, 18, 1829–1842. [DOI] [PubMed] [Google Scholar]
- Stamos J.; Sliwkowski M. X.; Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–272. [DOI] [PubMed] [Google Scholar]
- Homology model was built using the PRIME homology modeling program. Prime, 1.0 ed.; Schrodinger, Inc. Compound 2 has been docked manually by analogy to the docking mode of Erlotinib inhibitor in the reference structure 1M17. Images in Figure 2 have been created in VIDA v4 from OpenEye Scientific Software, 3600 Cerrillos Rd., Suite 1107, Santa Fe, NM 87507.
- Allen F. H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. 2002, B58, 380–388. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PDB code 3CGO:Buckley G. M.; Ceska T. A.; Fraser J. L.; Gowers L.; Groom C. R.; Higueruelo A. P.; Jenkins K.; Mack S. R.; Morgan T.; Parry D. M.; Pitt W. R.; Rausch O.; Richard M. D.; Sabin V. IRAK-4 inhibitors. Part II: a structure-based assessment of imidazo[1,2-a]pyridine binding. Bioorg. Med. Chem. Lett. 2008, 18, 3291–3295. [DOI] [PubMed] [Google Scholar]
- PDB code 2B55:Yue E. W.; Higley C. A.; Dimeo S. V.; Carini D. J.; Nugiel D. A.; Benware C.; Benfield P. A.; Cox S.; Burton C. R.; Grafstrom R. H.; Sharp D. M.; Sisk L. M.; Boylan J. F.; Muckelbauer J. K.; Smallwood A. M.; Chen H.; Chang C.-H.; Seitz S. P.; Trainor G. L. Synthesis and evaluation of indenopyrazoles as cyclin-dependent kinase inhibitors. 3. Structure activity relationships at C3(1,2). J. Med. Chem. 2002, 45, 5233–5248. [DOI] [PubMed] [Google Scholar]
- PDB code 2R4B:Wood E. R.; Shewchuk L. M.; Ellis B.; Brignola P.; Brashear R. L.; Caferro T. R.; Dickerson S. H.; Dickson H. D.; Donaldson K. H.; Gaul M.; Griffin R. J.; Hassell A. M.; Keith B.; Mullin R.; Petrov K. G.; Reno M. J.; Rusnak D. W.; Tadepalli S. M.; Ulrich J. C.; Wagner C. D.; Vanderwall D. E.; Waterson A. G.; Williams J. D.; White W. L.; Uehling D. E. 6-Ethynylthieno[3,2-d]- and 6-ethynylthieno[2,3-d]pyrimidin-4-anilines as tunable covalent modifiers of ErbB kinases. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 2773–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.