

Abstract
Semisynthetic analogues of fumagillin, 1, inhibit methionine aminopeptidase-2 (MetAP2) and have entered the clinic for the treatment of cancer. An optimized fumagillin analogue, 3 (PPI-2458), was found to be orally active, despite containing a spiroepoxide function that formed a covalent linkage to the target protein. In aqueous acid, 3 underwent ring-opening addition of water and HCl, leading to four products, 4–7, which were characterized in detail. The chlorohydrin, but not the diol, products inhibited MetAP2 under weakly basic conditions, suggesting reversion to epoxide as a step in the mechanism. In agreement, chlorohydrin 6 was shown to revert rapidly to 3 in rat plasma. In an ex vivo assay, rats treated with purified acid degradants demonstrated inhibition of MetAP2 that correlated with the biochemical activity of the compounds. Taken together, the results indicate that degradation of the parent compound was compensated by the formation of active equivalents leading to a pharmacologically useful level of MetAP2 inhibition.
Keywords: Fumagillin, MetAP2, PPI-2458, angiogenesis inhibitor, irreversible inhibitor
Drugs that contain reactive functional groups (e.g., Michael acceptors, epoxides, and β-lactams) and that inhibit their molecular targets by forming stable covalent linkages represent extreme cases of slow off-rate binding, where drug occupancy may become governed by protein turnover or other cellular processes that occur on an extended time scale.1 A particular advantage of such drugs is that provided the on rate is sufficiently rapid, a pharmacological effect may be achieved even without high or continuous exposure. Because of their reactivity, irreversible inhibitors are often thought to carry an enhanced risk of off-target toxicity, for example, by haptenization of proteins leading to an autoimmune response. Although side effects have been associated with well-known drugs of this type, notably allergic responses to penicillins, few cases of mechanism-related toxicity have been established in detail.2 Inclusion of in vivo testing at an early stage in the discovery process can help ensure that a drug candidate acting through an irreversible mechanism meets appropriate specificity and safety criteria. Another potential liability of this drug class results from in vivo processes that may degrade the warhead before it reaches its target. Specialized formulations, prodrugs, or nonoral routes of administration may be required to ensure bioavailability. Despite these challenges, drugs that act through a covalent mechanism have been approved for a diverse spectrum of diseases over the years, and a recent survey indicates a healthy pipeline of such molecules in the clinic.1
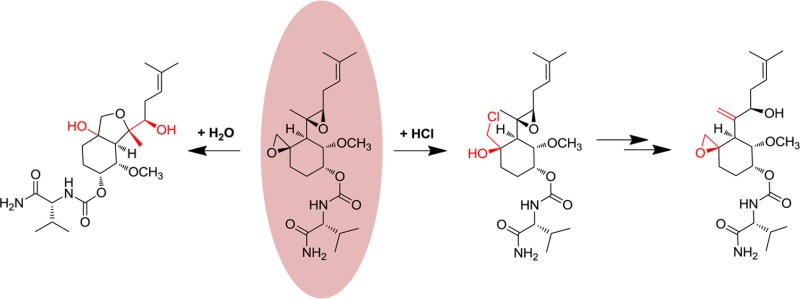
The sesquiterpenoid fumagillin 1 (Scheme 1) contains a spiroepoxide moiety that forms a covalent linkage with an active site histidine residue in the enzyme methionine aminopeptidase-2 (MetAP2).3 Serendipitously, the scaffold of this natural product, in particular the C4 side chain, mimics the N-methionyl residue of polypeptide chains that makes up the endogenous substrate of MetAP2.4 Noncovalent interactions arising from active site complementarity appear to play an important role in the effectiveness of fumagillin as an inhibitor: reversible fumagillin derivatives have been shown to inhibit MetAP2,5 whereas the simple cyclohexane spiroepoxide core is inactive.6 Long used as a veterinary antibiotic, fumagillin has attracted additional interest over the past 20 years because of its antiangiogenic effects.7 In particular, semisynthetic analogue 2 (TNP-470),8 in which the polyolefinic chain at C6 was truncated, was efficacious in animal models for cancer9 and entered human clinical trials.10 More recently, we disclosed compound 3 {(3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methylbut-2-en-1-yl)oxiran-2-yl]-1-oxaspiro[2.5]octan-6-yl [(R)-1-amino-3-methyl-1-oxobutan-2-yl]carbamate, PPI-2458},6 in which replacement of the chloroacetimide of 2 resulted in improved safety and pharmacokinetic properties, in particular oral bioavailability and a lack of CNS effects.11 Further studies demonstrated the utility of 3 in rodents as a disease-modifying agent for the treatment of rheumatoid arthritis.11−17 While highly desirable for treating chronic disease, the oral activity of 3 was surprising given the expected acid lability of the epoxide groups. Here, we describe the behavior of 3 in acidic solutions as a model for the gastric environment. The major products of acidic degradation were identified and characterized with respect to their ability to inhibit MetAP2, delineating a mechanism for the oral activity of 3.
Scheme 1. Structures of Compounds 1–3, with C4 and C6 Side Chains Marked.

Treatment of 3 with 6 mM HCl (pH 2) resulted in three products (4–6) that could be resolved by HPLC. The early eluting peaks corresponding to 4 and 5 showed a mass increase of 18 amu (+H2O), whereas the late eluting peak due to 6 displayed a mass increase of 36 amu (+HCl, confirmed from the 37Cl isotopomer). In simulated gastric fluid [0.7% (v/v) HCl and 0.2% NaCl (pH 1.2)], additional species were evident. In particular, another HCl adduct, 7, could be discerned eluting near 3 at 1.75 min. Over a 20 h time span, the quantity of 7 gradually increased relative to the quantity of 6 (Figure 1b).
Figure 1.

(a) Acid degradation of 3 in 6 mM HCl (pH 2). (b) Conversion of 6 (retention time of 2.14 min) to 7 (retention time of 1.75 min) at pH 1.
On a preparative scale, compounds 4 and 5 were synthesized by treatment of 3 with aqueous HCl, whereas a more convenient route to 6 and 7 was found using anhydrous HCl in dioxane. Under these conditions, the formation of 7 was rapid, and trapping 6 in useful quantities required neutralization of the reaction mixture after 5 min at room temperature. Both 6 and 7 reacted with KOtBu with loss of HCl, again yielding products that could be resolved by HPLC. In the case of 6, this material was isolated and shown to be identical with 3; the isomer formed from 7 was identified as a new compound, 8. Isolated compounds 4–8 were characterized by NMR spectroscopy, using previous analyses of 3 as a reference. Proton assignments were made using two-dimensional COSY, TOCSY, and ROESY correlations and are summarized in Table S1 of the Supporting Information.
As was the case with their HPLC retention times, the 1H NMR spectra of 4 and 5 were similar, but markedly different from that of 3. Notably, the resonance of H17, whose anomalously upfield chemical shift was interpreted in terms of its proximity to the spiroepoxide function, migrated downfield into the region of H14–H16. Significant chemical shift changes were also apparent for H6, H8, and H9. These changes were consistent with opening of the spiroepoxide ring. Oxidation of 4 and 5 with TPAP resulted in a loss of 2 amu and the disappearance of H6 as opposed to H8 or H9. Taken together, the data suggested that 4 and 5 could be assigned to isomeric perhydrobenzofuran structures (Scheme 2). In accordance with the guidelines for ring closure, the oxidation data excluded the alternative formation of a six-membered ring, which would have generated a tertiary alcohol.18 The absolute stereochemistry of both 4 and 5 was confirmed by crystallography (details in the Supporting Information), revealing the compounds as epimers at C3 (Figure 2). A plausible mechanism leading to 4 and 5 involves SN1 opening of the spiroepoxide and formation of diastereomeric diols, followed by SN2-type ring formation (Scheme 2).
Scheme 2. Summary of Structures and Pathways for Formation of Compounds 4–8.

Figure 2.
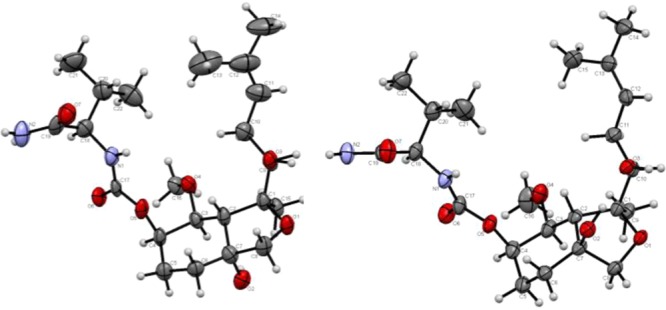
ORTEP plots of one molecule of 4 (left) and 5 (right). Displacement parameters are drawn at the 50% occupancy level.
The NMR spectrum of compound 6 was characterized by large changes in the chemical shifts of H6, Me7, H8, and H9, all of which moved downfield at least 0.4 ppm from their locations in the spectrum of 3. Together with the facile reversion of 6 to 3, the data support assignment of 6 as a chlorohydrin (Scheme 2). The 1H NMR spectrum of its isomer, 7, was quite distinct from that of 6, the most striking difference being the disappearance of the signal from Me7 and the emergence of a two-proton olefinic resonance. A weak coupling is seen between this peak and H6 in the TOCSY experiment, and a through-space correlation is observed to H4/5 in the ROESY experiment, supporting its assignment as the methylene formed by ring opening of the side chain epoxide. Finally, while the 1H NMR spectrum of 8 still exhibited the signature of the allylic alcohol in 7, other signals characteristic of 3, such as the upfield-shifted resonance of H17, were visible. This was consistent with re-formation of the spiroepoxide functionality in 8. Taken together, the data suggest that near pH 2, degradation proceeds through limited pathways featuring rapid opening of the more labile spiroepoxide by two distinct mechanisms for water and HCl. At lower pH values, processes involving the C4 side chain epoxide also contribute, leading to additional products.
Compounds 4–8 were tested for their ability to inhibit MetAP2 and HUVEC proliferation (Table 1).6 The enzyme assay monitors loss of MetAP2 activity with Met-AMC substrate over an 8 h window following treatment with a 10-fold excess of compound (200 nM), whereas the HUVEC assay records endothelial cell growth over 3 days in the presence of 0.02 nM to 2 μM compound. In the MetAP2 assay, compounds 4 and 5 failed to show measurable inhibition of MetAP2 over an 8 h time period and likewise did not attenuate growth of HUVEC cells at up to micromolar concentrations. Chlorohydrins 6 and 7, on the other hand, were active in both assays, as was epoxide 8. MetAP2 and HUVEC inhibition were correlated, with 6 nearly as efficacious as 3, whereas 7 and 8 were somewhat less active. The results underscore the importance of C4 side chain structure, insofar as comparatively minor changes resulting from the opening of the side chain epoxide in 7 and 8 can be accommodated, whereas the major rearrangement yielding the bicyclic structures of 4 and 5 is not compatible with the active site of MetAP2.
Table 1. Inhibition of MetAP2 Activity and HUVEC Proliferation by Compounds 3–8.
| compd | MetAP2 (% remaining activity)a | HUVEC IC50 (nM) |
|---|---|---|
| 3 | 3 ± 3 | 0.18 |
| 4 | 107 ± 6 | >30000 |
| 5 | 105 ± 6 | >30000 |
| 6 | 6 ± 2 | 0.15 |
| 7 | 58 | 3.3 |
| 8 | 27 | 3.1 |
Presented as residual MetAP2 activity after treatment with the compound for 8 h.
The similarity between the activity of the two spiroepoxides and their corresponding chlorohydrins prompted a closer study of the inhibitory mechanism. Progress curves for the inhibition of MetAP2 in HEPES buffer (pH 7.5) showed rapid inactivation by 3, whereas that of 6 proceeded more gradually (Figure 3a). When isomeric species 7 and 8 were also considered, the rates of inhibition decreased in the following order: 3 > 6 > 8 > 7 (not shown). The activity of 6 was also highly pH sensitive: in MES buffer (pH 6.5), no inhibition was observed, whereas 3 was equally effective under both conditions (Figure 3b). This behavior is consistent with a mechanism in which the chlorohydrins lack intrinsic activity but revert to epoxides under slightly basic conditions as an obligatory step toward inhibition. In agreement, we determined that 6 was ∼50% converted to 3 after 8 h in HEPES but was largely unchanged in MES. Because no reversion from 8 to 3 has been observed, we conclude that 8 is a direct, albeit less optimal, inhibitor of MetAP2.
Figure 3.
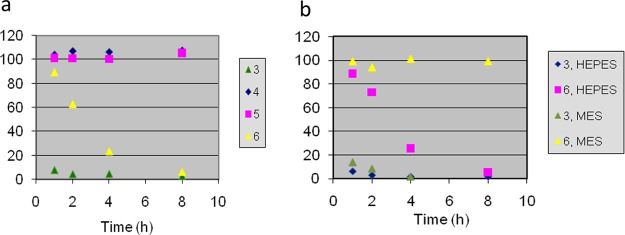
Time dependence of MetAP2 activity (% relative to DMSO or ethanol control) in the presence of (a) compounds 3–6 in HEPES (pH 7.5) and (b) 3 and 6 in HEPES (pH 7.5) or MES (pH 6.5).
Separately, we tested the stability of 6 in rat plasma. Exposure for 1 h at 4 °C resulted in quantitative reversion to 3 (Table S2 of the Supporting Information), whereas the amount in solvent controls is negligible. Close to 10% reversion occurs within the first minute. Thus, the kinetics of reversion of 6 to 3 in plasma greatly exceeds that observed in simple pH 7.5 buffer, possibly because of protein binding. One may surmise that in vivo, active species (3 and probably 8) are efficiently regenerated upon absorption from the gut.
Finally, we investigated the ability of the purified degradants to inhibit MetAP2 in vivo. Following oral dosing in rats, cells from whole blood, liver, and thymus were harvested and treated with a biotinylated form of 3.11 Free MetAP2 was thus labeled with an affinity tag, captured on streptavidin beads, and quantitated in an ELISA format. As shown in Figure 4, a single dose of 3 at 3 mg/kg effectively inhibited MetAP2 in all three tissues, whereas perhydrobenzofurans 4 and 5 were inactive. Compounds 6–8, while less effective than 3, nonetheless significantly inhibited MetAP2 in all compartments studied. Thus, the level of MetAP2 inhibition in vivo tracks potency in the biochemical assay. Notably, compounds 3 and 6–8 had reduced activity in solid tissues, including thymus, with a disparate drop in activity noted with compounds 6 and 7. While tissue concentrations were not determined, these findings may suggest that the distribution of these compounds into tissues may be variable compared to that in plasma. Plasma analysis following dosing with purified degradants confirmed the presence of 4, 5, and 8 in vivo, with measurements at 2 and 24 h indicating that their exposure levels matched or exceeded that of 3 when administered at an equal dose (Table S3 of the Supporting Information). Because the LC–MS/MS protocol specifically quantitated levels of the parent molecule, the exposure to active species (which in the case of 3 could include 8, as well as cytochrome P450-derived metabolites) is probably underestimated.
Figure 4.
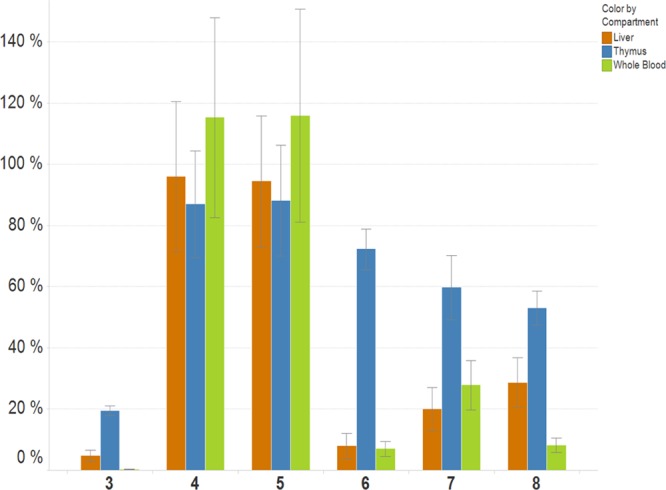
Free MetAP2 levels in liver, thymus, and whole blood following a single PO (3 mg/kg) dose of 3–8 as a percentage of vehicle control. Data are averages of triplicate experiments with percentage uncertainties shown.
Previous work has demonstrated some tolerance for modifications to the C4 side chain that are compatible with MetAP2 inhibition. CYP3A metabolism of 3 yields diastereomeric triepoxides as well as an allylic alcohol that contribute significantly to MetAP2 inhibition in vivo.19 While most drug discovery efforts have focused on other regions of the molecule, in particular the C6 chain, a highly potent inhibitor in which the C4 chain is replaced by a benzyl oxime has been reported by Pyun et al.20 Compounds 4 and 5 also have precedent, beginning with Tarbell’s extensive studies on fumagillin chemistry.21 More recently, an analogous derivative of fumagillin (fumagiringillin) was isolated from the fungus Aspergillus fumigatus, apparently as a single diastereomer.22 Studies of the earlier semisynthetic drug 2, which is rapidly metabolized in vivo, identified a perhydrobenzofuran as the major product of the metabolic pathway.23
The results presented here, together with findings from CYP metabolism studies, contribute to a detailed picture of the ADME properties of 3.24 In an acidic milieu modeling the environment of the stomach, two of the four major degradants retain MetAP2 inhibitory activity and have been characterized as ring-opened chlorohydrins. Animals orally dosed with parent compound 3, its isomer 8, or the corresponding chlorohydrins 6 and 7 exhibit significant inhibition of MetAP2 in blood and tissue. The degree of inhibition correlates with the biochemical efficacy of each species, with allylic alcohols 7 and 8 showing an overall reduction in potency relative to 3 and 6. Activity in vivo appears to require re-formation of the spiroepoxide, which is likely to occur upon passage into the neutral environment of the gut, or immediately upon absorption into the bloodstream. Thus, among the inhibitory compounds, only parent molecule 3 and its isomer 8 are detectable in plasma. Loss of 3 to inactive species such as 4 and 5 is compensated by its irreversible mechanism of action. The correlation of in vitro and in vivo SAR highlights the specific nature of MetAP2 inhibition by 3, providing support for its utility as a therapeutic candidate.
Acknowledgments
We thank Drs. Roy Copley and Randy Rutkowske of GlaxoSmithKline for careful review of the crystallography data and for obtaining the 13C NMR spectra, respectively.
Glossary
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion
- AMC
7-amino-4-methylcoumarin
- BLQ
below limit of quantitation
- CNS
central nervous system
- CYP
cytochrome P450
- DIEA
diisopropylethylamine
- DQF-COSY
double-quantum-filtered correlated spectroscopy
- ELISA
enzyme-linked immunosorbent assay
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPCD
hydroxypropylcyclodextrin
- HUVEC
human umbilical vascular endothelial cell
- IV
intravenous
- MES
2-(N-morpholino)ethanesulfonic acid
- MetAP2
methionine aminopeptidase-2
- PO
orally dosed
- ROESY
rotating frame Overhauser spectroscopy
- TOCSY
total correlation spectroscopy
- TPAP
tetrapropylammonium perruthenate
- WFI
water for injection.
Supporting Information Available
Procedures for the synthesis of compounds 4–8, NMR resonance assignments, details of the crystal structures of compounds 4 and 5 (with CIF files), procedures for biochemical and the HUVEC assay, procedure for testing the plasma stability of 6, and the protocol for measuring plasma levels and MetAP2 inhibition in rats. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† GlaxoSmithKline, 830 Winter St., Waltham, MA 02451–1420.
Author Present Address
‡ Cubist Pharmaceuticals, 65 Hayden Ave., Lexington, MA 02421.
Author Present Address
§ Seattle Genetics, Inc., 21823 30th Dr. SE, Bothell, WA 98021.
Author Present Address
∥ Pfizer Inc., One Burtt Rd., Andover, MA 01810.
Author Present Address
⊥ X-Chem, Inc., 100 Beaver St., Waltham, MA 02453.
Author Present Address
@ 175 Oakdale St., Attleboro, MA 02703.
Author Present Address
# Vertex Pharmaceuticals, 130 Waverly St., Cambridge, MA 02139.
Author Present Address
∇ Rutgers, The State University of New Jersey, Mario School of Pharmacy, 160 Frelinghuysen Rd., Piscataway, NJ 08854.
Author Present Address
○ Celgene Avilomics Research, 45 Wiggins Ave., Bedford, MA 01730.
Author Present Address
● 237 Prospect St., Franklin, MA 02038.
Author Present Address
◆ Department of Chemistry, Northwestern University, 2145 Sheridan Rd., Evanston, IL 60208.
Author Present Address
◇ Merck & Co. Inc., West Point, PA 19486.
Author Present Address
▲ Chemical Research and Development, Pfizer, Eastern Point Road, Groton, CT 06340.
Author Present Address
△ Infinity Pharmaceuticals, 780 Memorial Dr., Cambridge, MA 02139.
The authors declare the following competing financial interest(s): C.C.A.-M., G.O., and S.S. are employees of GlaxoSmithKline, which now owns rights to PPI-2458.
Supplementary Material
References
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- Potashman M. H.; Duggan M. E. Covalent modifiers: An orthogonal approach to drug design. J. Med. Chem. 2009, 52, 1231–1246. [DOI] [PubMed] [Google Scholar]
- Griffith E. C.; Su Z.; Niwayama S.; Ramsay C. A.; Chang Y.-H.; Liu J. O. Molecular recognition of angiogenesis inhibitors fumagillin and ovalicin by methionine aminopeptidase 2. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 15183–15188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Widom J.; Kemp C. W.; Crews C. M.; Clardy J. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science 1998, 282, 1324–1327. [DOI] [PubMed] [Google Scholar]
- Zhou G.; Tsai C. W.; Liu J. O. Fumagalone, a reversible inhibitor of type 2 methionine aminopeptidase and angiogenesis. J. Med. Chem. 2003, 46, 3452–3454. [DOI] [PubMed] [Google Scholar]
- Arico-Muendel C. C.; Benjamin D. R.; Caiazzo T. M.; Centrella P. A.; Contonio B. D.; Cook C. M.; Doyle E. G.; Hannig G.; Labenski M. T.; Searle L. L.; Lind K.; Morgan B. A.; Olson G.; Paradise C. L.; Self C.; Skinner S. R.; Sluboski B.; Svendsen J. L.; Thompson C. D.; Westlin W.; White K. F. Carbamate analogues of fumagillin as potent, targeted inhibitors of methionine aminopeptidase-2. J. Med. Chem. 2009, 52, 8047–8056. [DOI] [PubMed] [Google Scholar]
- Ingber D.; Fujita T.; Kishimoto S.; Sudo K.; Kanamaru T.; Brem H.; Folkman J. Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumor growth. Nature 1990, 348, 555–557. [DOI] [PubMed] [Google Scholar]
- Marui S.; Itoh F.; Kozai Y.; Sudo K.; Kishimoto S. Chemical modification of fumagillin. I. 6-O-Acyl, 6-O-Alkyl, and 6-O-(N-Substituted-carbamoyl)fumagillols. Chem. Pharm. Bull. 1992, 40, 96–101. [DOI] [PubMed] [Google Scholar]
- Folkman J.Tumor angiogenesis. In Accomplishments in Cancer Research 1997; Wells S. A., Sharp P. A., Eds.; Lippincott Williams & Wilkins: Philadelphia, 1998; pp 32–44. [Google Scholar]
- Bernier S. G.; Westlin W. F.; Hannig G. Fumagillin class inhibitors of methionine aminopeptidase-2. Drugs Future 2005, 30, 497–508. [Google Scholar]
- Bernier S. G.; Lazarus D. D.; Clark E.; Doyle B.; Labenski M. T.; Thompson C. D.; Westlin W. F.; Hannig G. A methionine aminopeptidase-2 inhibitor, PPI-2458, for the treatment of rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 10768–10773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannig G.; Bernier S. G.; Hoyt J. G.; Doyle B.; Clark E.; Karp R. M.; Lorusso J.; Westlin W. F. Suppression of inflammation and structural damage in experimental arthritis through molecular targeted therapy with PPI-2458. Arthritis Rheum. 2007, 56, 850–860. [DOI] [PubMed] [Google Scholar]
- Lazarus D. D.; Doyle E. G.; Bernier S. G.; Rogers A. B.; Labenski M. T.; Wakefield J. D.; Karp R. M.; Clark E. J.; Lorusso J.; Hoyt J. G.; Thompson C. D.; Hannig G.; Westlin W. F. An inhibitor of methionine aminopeptidase type-2, PPI-2458, ameliorates the pathophysiological disease processes of rheumatoid arthritis. Inflammation Res. 2008, 57, 18–27. [DOI] [PubMed] [Google Scholar]
- Bainbridge J.; Madden L.; Essex D.; Binks M.; Malhotra R.; Paleolog E. M. Methionine aminopeptidase-2 blockade reduces chronic collagen-induced arthritis: Potential role for angiogenesis inhibition. Arthritis Res. Ther. 2007, 9, R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahn E.; Schoettler N.; Lee S.; Banquerigo M. L. Involution of collagen-induced arthritis with an angiogenesis inhibitor, PPI-2458. J. Pharm. Exp. Ther. 2009, 329, 615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf S.; Mapp P. I.; Walsh D. A. Angiogenesis and the persistence of inflammation in a rat model of proliferative synovitis. Arthritis Rheum. 2010, 62, 1890–1898. [DOI] [PubMed] [Google Scholar]
- Ashraf S.; Mapp P. I.; Walsh D. A. Contributions of angiogenesis to inflammation, joint damage, and pain in a rat model of osteoarthritis. Arthritis Rheum. 2011, 63, 2700–2710. [DOI] [PubMed] [Google Scholar]
- Baldwin J. E. Rules for ring closure. J. Chem. Soc., Chem. Commun. 1976, 18, 734–736. [Google Scholar]
- Thompson C.; Arico-Muendel C.. Fumagillin-related compound inhibitors of methionine aminopeptidase 2 and use for the treatment of angiogenic and other conditions. WO 2005066197, 2005.
- Pyun H.-J.; Fardis M.; Tario J.; Yang C. Y.; Ruckman J.; Henninger D.; Jin H.; Kim C. U. Investigation of novel fumagillin analogues as angiogenesis inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 91–94. [DOI] [PubMed] [Google Scholar]
- Tarbell D. S.; Carman R. M.; Chapman D. D.; Cremer S. E.; Cross A. D.; Huffman K. R.; Kunstmann M.; McCorkindale N. J.; McNally J. G. Jr.; Rosowsky A.; Varino F. H. L.; West R. L. The chemistry of fumagillin. J. Am. Chem. Soc. 1961, 83, 3096–3113. [Google Scholar]
- Jiao W.; Blunt J. W.; Cole A. L. J.; Munroe M. H. G. Fumagiringillin, a new fumagillin derivative from a strain of the fungus Aspergillus fumigatus. J. Nat. Prod. 2004, 67, 1434–1437. [DOI] [PubMed] [Google Scholar]
- Placidi L.; Cretton-Scott E.; De Sousa G.; Rahmani R.; Placidi M.; Sommadossi J.-P. Disposition and metabolism of the angiogenic moderator O-(chloroacetyl-carbamoyl) fumagillol (TNP-470; AGM-1470) in human hepatocytes and tissue microsomes. Cancer Res. 1995, 55, 3036–3042. [PubMed] [Google Scholar]
- Arico-Muendel C. C.; Belanger B.; Benjamin D.; Blanchette H. S.; Caiazzo T. M.; Centrella P. A.; DeLorey J.; Doyle E. G.; Gradhand U.; Griffin S. T.; Hill S.; Labensky M. T.; Morgan B. A.; O’Donovan G.; Prasad K.; Skinner S.; Taghizadeh N.; Thompson C.; Wakefield J.; Westlin W.; White K. F. Metabolites of PPI-2458, a selective, irreversible inhibitor of methionine aminopeptidase-2. Structure determination and in vivo activity. Drug Metab. Dispos. 2013, DOI: 10.1124/dmd.112.048355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.