

Abstract
Aberrant activation of the Wnt pathway has been implicated in the development and formation of many cancers. TNKS inhibition has been shown to antagonize Wnt signaling via Axin stabilization in APC mutant colon cancer cell lines. We employed structure-based design to identify a series of 2-aminopyridine oxazolidinones as potent and selective TNKS inhibitors. These compounds exhibited good enzyme and cell potency as well as selectivity over other PARP isoforms. Co-crystal structures of these 2-aminopyridine oxazolidinones complexed to TNKS reveal an induced-pocket binding mode that does not involve interactions with the nicotinamide binding pocket. Oral dosing of lead compounds 3 and 4 resulted in significant effects on several Wnt-pathway biomarkers in a three day DLD-1 mouse tumor PD model.
Keywords: Wnt pathway, tankyrase inhibitor, cancer, PARP, 2-aminopyridine oxazolidinone
The canonical Wnt pathway regulates the stability of the transcriptional coactivator β-catenin.1 The level of active β-catenin is tightly controlled by a complex of proteins known as the destruction complex, which consists of the scaffolding protein Axin, kinases GSK3β and CK1α, and the tumor suppressor protein APC.2 In the absence of Wnt stimulation, cytosolic β-catenin is constitutively phosphorylated and targeted for degradation by the ubiquitin/proteosome pathway. Interaction of a Wnt ligand with cell surface receptors results in breakdown of the β-catenin destruction complex, leading to the accumulation of nuclear β-catenin and transcription of Wnt target genes.3,4
Aberrant activation of the Wnt pathway has been implicated in the development and formation of many cancers; for example, APC mutations have been observed in >80% of sporadic colorectal cancers (CRC).5,6 Blocking the aberrant Wnt signaling has been shown to be effective in stopping growth of cultured colon cancer cells.7,8 Considering the increasing number of Wnt-driven cancers, intervention of the Wnt signaling pathway with small molecules has the potential to be an effective cancer treatment.9−11
Tankyrases belong to the poly-ADP-ribose polymerase (PARP) family.12 The two isoforms, TNKS1 and TNKS2, share overlapping function and a high degree of homology. Seminal work by researchers at Novartis and University of Texas, Southwestern demonstrated that pharmacological inhibition of TNKS1/2 antagonizes canonical Wnt signaling via Axin stabilization in certain APC mutant colon cancer cell lines (SW480, DLD-1).13,14 In this research, two main structural classes of small molecule TNKS inhibitors were utilized: those that occupy the highly conserved nicotinamide pocket of the catalytic PARP domain, as represented by XAV939,15 and those that bind to the induced pocket of the enzymes, as exemplified by IWR1/2.16,17 The induced pocket is not present in apo TNKS proteins and only becomes accessible upon the binding of the inhibitors.18 Residues forming the induced pocket are less conserved among other PARP family members. Therefore, this novel binding mode provides a unique opportunity to achieve broader selectivity over other PARP family members and NAD-utilizing enzymes.19,20 Herein, we describe a structure-based approach to identify novel 2-aminopyridine oxazolidinones as potent and selective TNKS inhibitors that bind in the induced pocket.
The compounds presented in this study were examined for their ability to inhibit the activity of TNKS1 using enzyme assays with recombinant TNKS catalytic domains. Three cellular assays in two different APC mutant CRC cell lines were developed to aid evaluation of TNKS inhibitors. First, a proximal cellular assay measured the accumulation of Axin2 protein in SW480 cells as a result of TNKS inhibition. Second, a central cellular assay assessed the degradation of total β-catenin (TBC) in SW480 cells. Finally, a transcriptional Wnt-reporter assay (STF), driven by multimerized TCF binding sites in DLD-1 cells quantified the effect of TNKS inhibition on downstream Wnt signaling events.
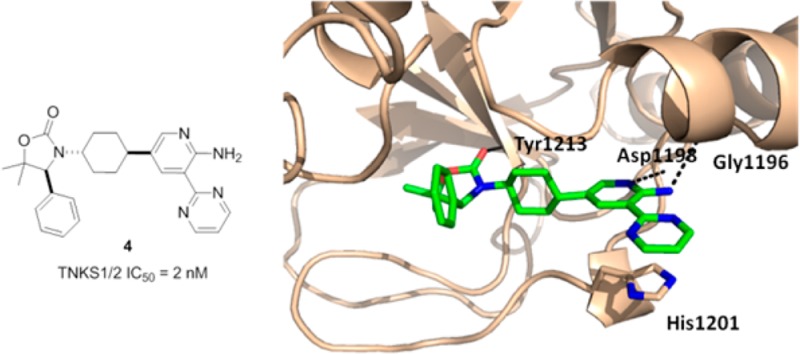
Recently we discovered that oxazolidinone 1 binds to TNKS1 in the induced pocket in a manner similar to that described for IWR1/2 (Figure 1A).21 The key ligand–protein interactions include three hydrogen bonds: the carbonyl group of the oxazolidinone interacts with Tyr1213; the carbonyl group of the amide with Asp1198; the CH at the 6-position of the quinoline is associated with the backbone carbonyl of Gly1196. In addition, the quinoline group engages in a π-stacking interaction with His1201 of the D-loop (Figure 1B). While 1 showed good enzymatic activity, it exhibited moderate rat plasma stability. Metabolite identification studies revealed that the amide is prone to hydrolysis in both rat and mouse plasma.
Figure 1.

(A) Structure and activity of compounds 1 and 2. (B) Cocrystal structure of TNKS1 complexed with 1. (C) Cocrystal structure of TNKS1 complexed with 2. (D) Overlay of cocrystal structures of 1 and 2.
During the course of our lead-optimization work on 1, high-throughput screening identified compound 2 as a potent and novel TNKS inhibitor. A cocrystal structure of 2 complexed with TNKS1 reveals that 2 occupies both the conserved nicotinamide pocket and the induced pocket in a dual-binding mode, similar to that recently described for other TNKS inhibitors.22 The dihydroquinolinone moiety resides in the nicotinamide pocket with the carbonyl group hydrogen bonding to Ser1221 and the nitrogen engaging in a hydrogen bond interaction with Gly1185. The aminoquinazoline group occupies the induced pocket, with N1 hydrogen bonded to Asp1198, and the amino group hydrogen bonded to Gly1196 (Figure 1C).
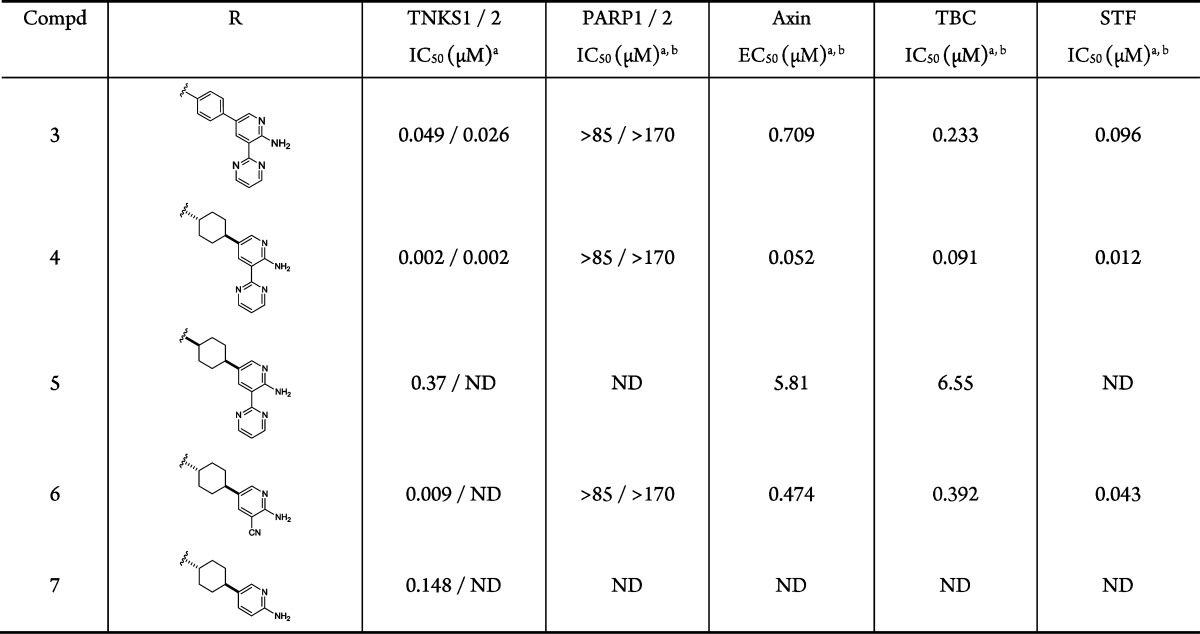
An overlay of the cocrystal structures of 1 and 2 bound to TNKS1 inspired a hypothesis that replacing the labile aminoquinazoline amide of 1 with a 2-aminopyridine might increase stability in plasma while maintaining two critical H-bond interactions with the protein (Figure 1A). We envisioned that the aminopyridine moiety would serve as a simplified mimic of the aminoquinazoline group in 2. Positioning an aromatic pyrimidinyl group at the 3-positon would engage the important π-stacking interaction with His1201, as observed with the quinoline ring of 1. Gratifyingly, compound 3 showed good TNKS inhibitory activity while demonstrating excellent selectivity over PARP1/2 (Table 1). In cellular assays, 3 promoted accumulation of Axin (Axin EC50 = 0.709 μM) and inhibited β-catenin accumulation (TBC IC50 = 0.233 μM) and Wnt-reporter gene transcription (STF IC50 = 0.096 μM). Furthermore, compound 3 was stable in human and rat liver microsomes (HLM/RLM Clint = 33/48 μL/min/mg).
Table 1. SAR of 2-Aminopyridine Oxazolidinones.

Values represent the mean of at least of two experiments.
ND = not determined.
Consistent with previously disclosed observations,13 potency was significantly improved by replacing the central phenyl ring with a saturated cyclohexyl group. The stereochemistry of the cyclohexyl ring significantly impacts the potency. For example, the trans-isomer 4 demonstrated greatly improved enzymatic and cellular potency over 3, while the cis-isomer 5 was less potent. The substituent at the 3-position of the pyridine ring proved to be important for potency. While compound 7 showed only moderate enzymatic activity, introduction of a small polar group (−CN) in compound 6 led to enhanced potencies in the enzyme and cellular assays. Although highly efficient, 6 had reduced potency relative to lead compound 4, which contains the aromatic pyrimidine substituent incorporated in the initial design for π-stacking to His1201.
To understand the structural basis for the potency observed with compounds 3 and 4, cocrystal structures of both compound 3 and 4 complexed with TNKS1 were obtained. The structures were resolved at 2.3 and 2.0 Å, respectively (Figure 2). The two compounds bind to TNKS1 in a similar fashion. The carbonyl group of the oxazolidinone is hydrogen bonded to Tyr1213. As intended, the aminopyridine interacts with Asp1198 and Gly1196 through two hydrogen bond interactions. The pyrimidine ring is close to be being coplanar with the aminopyridine ring and engages in a π-stacking interaction with His1201 of the D-loop. The major difference between the two structures is the position of the central ring of the inhibitors. The central phenyl ring of 3 has a ∼45 degree dihedral angle with respect to the pyridine ring; while the trans-cyclohexyl group of 4 is perpendicular to the pyridine ring, a conformation that enables the cyclohexyl group to fill the hydrophobic pockets formed by neighboring residues (Ser1186, Phe1188, and Ala1202). These hydrophobic interactions are not present in the 3/TNKS1 structure, which may account for the increased potency of 4 relative to 3.
Figure 2.
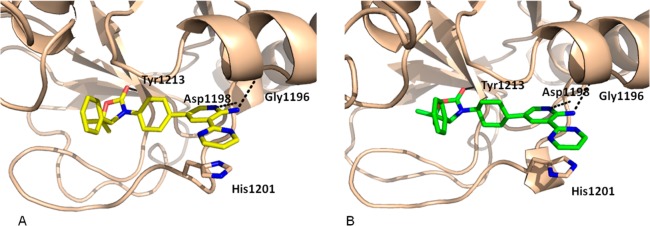
X-ray cocrystal structures: (A) 3 bound to TNKS1 and (B) 4 bound to TNKS1.
Since the initial lead 2 was a potent kinase inhibitor and the 2-aminopyridine motif is known to interact with the hinge region of many kinases,23 we screened 3 and 4 against a broad panel of kinases (Ambit KINOMEscan).24 Both compounds exhibited weak kinase inhibitory activity in this panel. Only 1 out of the 100 kinases had percent-of-control (POC) < 30 at 1 μM of 4.25 Compounds 3 and 4 were also screened for genotoxicity potential. Intriguingly, while 3 tested positive in a microAmes assay with and without S9 activation, 4 was negative under the same assay conditions. Compound 4 is also highly selective over 13 GPCRs (EC50, IC50 > 48.5 μM),26 hERG (IC50 = 10.9 μM), and BSEP (IC50 = 11 μM).
In light of their excellent enzyme and cellular potencies and good selectivity, compounds 3 and 4 were evaluated in rodent pharmacokinetic (PK) studies. Selected PK data is summarized in Table 2. Upon intravenous (iv) dosing to rats, 3 and 4 demonstrated moderate clearance and volumes of distribution with elimination half-lives of 4.3 and 3.6 h, respectively. Both compounds exhibited reasonable plasma exposure and bioavailability when dosed orally in rats at 2.0 mg/kg. Oral dosing in mice at 30 mg/kg also resulted in a good plasma exposure.
Table 2. Pharmacokinetic Parameters of Compounds 3 and 4.
| compd | rat iv PKa |
rat po PKa |
mouse po PKb |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dosec (mg/kg) | CL (L/h/kg) | Vss (L/kg) | t1/2 (h) | dosed (mg/kg) | Cmax (μM) | t1/2 (h) | AUC (μM·h) | F (%) | dosee (mg/kg) | Cmax (μM) | t1/2 (h) | AUC (μM·h) | |
| 3 | 0.5 | 0.52 | 2.32 | 4.3 | 2.0 | 0.37 | 4.6 | 2.71 | 29 | 30 | 7.88 | 3.7 | 63.3 |
| 4 | 0.5 | 0.75 | 2.85 | 3.6 | 2.0 | 0.45 | 2.9 | 1.98 | 33 | 30 | 6.09 | 3.5 | 36.3 |
Pharmacokinetic parameters following administration of compounds 3 or 4 in male Sprague–Dawley rats: 2 animals per study.
Pharmacokinetic parameters following administration of compound 3 or 4 in male Mouse_CD1: 3 animals per study.
Dosed as a solution in DMSO.
Dosed in 1% Tween 80/2% HPMC/97% water/MSA pH 2.2.
Dosed in 10.0% Pluronic F68/30.0% HPBCD/60.0% water/MSA pH 2.5.
Compounds 3 and 4 were evaluated in a pharmacodynamic (PD) assay in DLD-1 human tumor xenograft mice. Oral b.i.d. dosing of 3 at 200 mg/kg for three days resulted in significant Axin accumulation and inhibition of STF reporter activity with associated terminal plasma concentration of approximately 22.1 μM (Figure 3). In addition, 3 also inhibited Wnt-target gene (Axin2 and TCF7) expression at the same dose (Supporting Information). More potent lead 4 promoted Axin accumulation and STF reporter activity inhibition at a lower dose (50 mg/kg, oral b.i.d.) with associated terminal plasma concentration of 0.12 μM.
Figure 3.
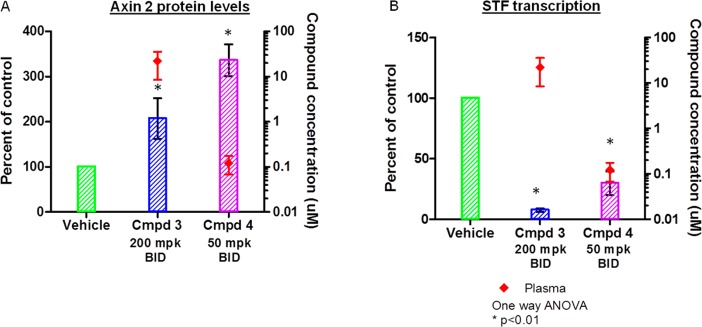
Subcutaneous tumors were established in athymic nude mice using DLD-1 human colon cancer cell line. Animals were dosed for 3 days and then tumors (for protein analysis) and plasma (for PK) were collected 14 h after the last dose. (A) The effect of compounds 3 and 4 on accumulation of Axin2 protein in human tumor xenograft model. (B) The effect of compounds 3 and 4 on inhibition of downstream reporter gene transcription.
In order to ascertain general utility for extended pharmacodynamic studies, compound 4 was dosed orally for eight days to naïve athymic nude mice. At 50 mg/kg b.i.d. continuous dosing, compound 4 was well tolerated. This is encouraging as this was the efficacious dose in the DLD-1 tumor PD study, and the mean unbound plasma concentration levels were comparable between the two studies.27
Despite robust in vivo Axin accumulation and inhibition of STF in a three day DLD-1 mouse tumor xenograft assay, compounds 3 and 4 were not advanced to multiweek DLD-1 tumor xenograft efficacy studies because the observed cell-killing activity of 4 in a standard three day DLD-1 cell proliferation assay (10% FBS, nuclear counting with cellomics Array scan) was only moderate (IC50 = 3 μM), and recent data suggests that DLD-1 tumor growth is not sensitive to pharmacological TNKS inhibition.28
Compound 4 is an optimized representative of this highly potent and selective structural series with potency, selectivity, and pharmacokinetic properties suitable to explore the pharmacological potential of TNKS inhibition in the context of future mouse tumor xenograft efficacy studies.
Compound 3 was prepared in a convergent fashion as described in Scheme 1. Suzuki coupling between pyridinyl boronic acid 8 and 2-bromopyrimidine afforded aryl bromide 9, which was subsequently converted to boronic acid pinacol ester 10 in good yield. A copper catalyzed coupling of commercially available 11 with 1,4-diiodobenzene provided aryl iodide 12. Suzuki coupling of 10 with 12 followed by aminolysis of the 2-fluoropyridine then furnished compound 3 in high yield.
Scheme 1. Synthesis of Compound 3.

Reagents: (a) 2-bromopyrimidine, PdCl2(dppf), aq. Na2CO3, dioxane, 100 °C, 76%; (b) PdCl2(dppf), KOAc, bis(pinacolato)diboron, 79%; (c) 1,4-diiodobenzene, CuI, K3PO4, MeNH(CH2)2NHMe, dioxane, μW, 140 °C, 61%; (d) PdCl2(dppf), aq. Na2CO3, dioxane, 100 °C, 62%; (e) NH3, DMSO, 83%.
Compounds 4–7 were prepared using the routes illustrated in Scheme 2. Reductive amination between commercial amino alcohol (S)-13 and 1,4-cyclohexanedione monoethylene acetal provided compound 14. Triphosgene-mediated cyclization and subsequent deprotection of the ketal using TFA afforded ketone 15 in 81% yield over 3 steps. Notably the chiral integrity remained intact during the process. Ketone 15 was converted to triflate 16 under standard conditions. Suzuki coupling of 16 with 2-aminopyridine-3-boronic acid pinacol ester followed by Pd/C catalyzed hydrogenation provided a mixture of cis- and trans-isomers of 7 (∼1:1 ratio). The two isomers were separated by HPLC. Bromination of 7 under standard condition afforded intermediate 17. Stille coupling of 17 with 2-(tributylstannyl)pyrimidine provided compound 4. Pd-catalyzed cyanation of 17 using Zn(CN)2 furnished compound 6.
Scheme 2. Synthesis of Compounds 4-7.

Reagents: (a) 1,4-cyclohexanedione monoethylene acetal, NaBH(OAc)3, DCM, rt; (b) triphosgene; (c) TFA, 81% (3 steps); (d) PhNTf2, LiHMDS, THF, 71%; (e) 2-aminopyridine-5-boronic acid pinacol ester, PdCl2(dppf), aq. Na2CO3, dioxane, 100 °C, 93%; (f) H2, Pd/C, 39%; (g) NBS, DCM, 100%; (h) 2-(tributylstannyl)pyrimidine, Pd(Ph3P)4, LiCl, CuI, DMF, 47%; (i) Pd(Ph3P)4, Zn(CN)2, DMF, 60%.
We have described the design, synthesis, and biological evaluation of a novel series of aminopyridine oxazolidinones as potent and selective TNKS inhibitors that bind in the induced pocket. Compound 4 demonstrated excellent TNKS potency in both enzyme and cell assays and exhibited high selectivity over PARP1/2. Twice daily oral dosing of 4 at 50 mg/kg promoted accumulation of Axin and inhibition of Wnt-dependent reporter activity in a three day DLD-1 mouse tumor PD model. This dosing schedule was also well tolerated in naïve athymic nude mice for eight continuous days. Compound 4 is expected to serve as an excellent tool compound to thoroughly investigate the effects of selective pharmacological inhibition of TNKS in various APC mutant CRC lines and disease relevant tumor models.
Glossary
ABBREVIATIONS
- APC
adenomatous polyposis coli
- CK1α
casein kinase 1α
- GSK3β
glycogen synthase kinase 3β
- STF
super TOP-flash
Supporting Information Available
Experimental details, biological assays, kinase selectivity, and X-ray crytstallographic information. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The PDB accession codes for the X-ray cocrystal structures of TNKS1 + 2, TNKS1 + 3, and TNKS1 + 4 are 4N3R, 4N4T, and 4N4V, respectively.
The authors declare no competing financial interest.
Supplementary Material
References
- Gordon M. D.; Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [DOI] [PubMed] [Google Scholar]
- Kimelman D.; Xu W. beta-Catenin destruction complex: insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [DOI] [PubMed] [Google Scholar]
- Yochum G. S.; Sherrick C. M.; Macpartlin M.; Goodman R. H. A beta-catenin/TCF-coordinated chromatin loop at MYC integrates 5′ and 3′ Wnt responsive enhancers. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirando A. J.; Maruyama T.; Fu J.; Yu H. M.; Hsu W. beta-Catenin/cyclin D1 mediated development of suture mesenchyme in calvarial morphogenesis. BMC Dev. Biol. 2010, 10, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V.; Barker N.; Morin P. J.; van Wichen D.; de Weger R.; Kinzler K. W.; Vogelstein B.; Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC–/– colon carcinoma. Science 1997, 275, 1784–1787. [DOI] [PubMed] [Google Scholar]
- Powell S. M.; Zilz N.; Beazer-Barclay Y.; Bryan T. M.; Hamilton S. R.; Thibodeau S. N.; Vogelstein B.; Kinzler K. W. APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [DOI] [PubMed] [Google Scholar]
- Tetsu O.; McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398, 422–426. [DOI] [PubMed] [Google Scholar]
- van de Wetering M.; Sancho E.; Verweij C.; de Lau W.; Oving I.; Hurlstone A.; van der Horn K.; Batlle E.; Coudreuse D.; Haramis A. P.; Tjon-Pon-Fong M.; Moerer P.; van den Born M.; Soete G.; Pals S.; Eilers M.; Medema R.; Clevers H. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [DOI] [PubMed] [Google Scholar]
- Huang H.; Acquaviva L.; Berry V.; Bregman H.; Chakka N.; O’Connor A.; Dimauro E. F.; Dovey J.; Epstein O.; Grubinska B.; Goldstein J.; Gunaydin H.; Hua Z.; Huang X.; Huang L.; Human J.; Long A.; Newcomb J.; Patel V. F.; Saffran D.; Serafino R.; Schneider S.; Strathdee C.; Tang J.; Turci S.; White R.; Yu V.; Zhao H.; Wilson C.; Martin M. W. Structure-based design of potent and selective CK1γ inhibitors. ACS Med. Chem. Lett. 2012, 3, 1059–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z.; Huang X.; Bregman H.; Chakka N.; Dimauro E. F.; Doherty E. M.; Goldstein J.; Gunaydin H.; Huang H.; Mercede S.; Newcomb J.; Patel V. F.; Turci S. M.; Yan J.; Wilson C.; Martin M. W. 2-Phenylamino-6-cyano-1H-benzimidazole-based isoform selective casein kinase 1 gamma (CK1gamma) inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5392–5395. [DOI] [PubMed] [Google Scholar]
- Waaler J.; Machon O.; Tumova L.; Dinh H.; Korinek V.; Wilson S. R.; Paulsen J. E.; Pedersen N. M.; Eide T. J.; Machonova O.; Gradl D.; Voronkov A.; von Kries J. P.; Krauss S. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012, 72, 2822–2832. [DOI] [PubMed] [Google Scholar]
- Lehtio L.; Chi N. W.; Krauss S. Tankyrases as drug targets. FEBS J. 2013, 280, 3576–3593. [DOI] [PubMed] [Google Scholar]
- Huang S. M.; Mishina Y. M.; Liu S.; Cheung A.; Stegmeier F.; Michaud G. A.; Charlat O.; Wiellette E.; Zhang Y.; Wiessner S.; Hild M.; Shi X.; Wilson C. J.; Mickanin C.; Myer V.; Fazal A.; Tomlinson R.; Serluca F.; Shao W.; Cheng H.; Shultz M.; Rau C.; Schirle M.; Schlegl J.; Ghidelli S.; Fawell S.; Lu C.; Curtis D.; Kirschner M. W.; Lengauer C.; Finan P. M.; Tallarico J. A.; Bouwmeester T.; Porter J. A.; Bauer A.; Cong F. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [DOI] [PubMed] [Google Scholar]
- Chen B.; Dodge M. E.; Tang W.; Lu J.; Ma Z.; Fan C. W.; Wei S.; Hao W.; Kilgore J.; Williams N. S.; Roth M. G.; Amatruda J. F.; Chen C.; Lum L. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlberg T.; Markova N.; Johansson I.; Hammarstrom M.; Schutz P.; Weigelt J.; Schuler H. Structural basis for the interaction between tankyrase-2 and a potent Wnt-signaling inhibitor. J. Med. Chem. 2010, 53, 5352–5355. [DOI] [PubMed] [Google Scholar]
- Narwal M.; Venkannagari H.; Lehtio L. Structural basis of selective inhibition of human tankyrases. J. Med. Chem. 2012, 55, 1360–1367. [DOI] [PubMed] [Google Scholar]
- Haikarainen T.; Venkannagari H.; Narwal M.; Obaji E.; Lee H. W.; Nkizinkiko Y.; Lehtio L. Structural basis and selectivity of tankyrase inhibition by a Wnt signaling inhibitor WIKI4. PLoS One 2013, 8, e65404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin H.; Gu Y.; Huang X. Novel binding mode of a potent and selective tankyrase inhibitor. PLoS One 2012, 7, e33740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund F. E. Signaling properties of CD38 in the mouse immune system: enzyme-dependent and -independent roles in immunity. Mol. Med. 2006, 12, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharehbaghi K.; Sreenath A.; Hao Z.; Paull K. D.; Szekeres T.; Cooney D. A.; Krohn K.; Jayaram H. N. Comparison of biochemical parameters of benzamide riboside, a new inhibitor of IMP dehydrogenase, with tiazofurin and selenazofurin. Biochem. Pharmacol. 1994, 48, 1413–1419. [DOI] [PubMed] [Google Scholar]
- Bregman H.; Chakka N.; Guzman-Perez A.; Gunaydin H.; Gu Y.; Huang X.; Berry V.; Liu J.; Teffera Y.; Huang L.; Egge B.; Mullady E. L.; Schneider S.; Andrews P. S.; Mishra A.; Newcomb J.; Serafino R.; Strathdee C. A.; Turci S. M.; Wilson C.; Dimauro E. F. Discovery of novel, induced-pocket binding oxazolidinones as potent, selective, and orally bioavailable tankyrase inhibitors. J. Med. Chem. 2013, 56, 4320–4342. [DOI] [PubMed] [Google Scholar]
- Bregman H.; Gunaydin H.; Gu Y.; Schneider S.; Wilson C.; DiMauro E. F.; Huang X. Discovery of a class of novel tankyrase inhibitors that bind to both the nicotinamide pocket and the induced pocket. J. Med. Chem. 2013, 56, 1341–1345. [DOI] [PubMed] [Google Scholar]
- Cui J. J.; Tran-Dube M.; Shen H.; Nambu M.; Kung P. P.; Pairish M.; Jia L.; Meng J.; Funk L.; Botrous I.; McTigue M.; Grodsky N.; Ryan K.; Padrique E.; Alton G.; Timofeevski S.; Yamazaki S.; Li Q.; Zou H.; Christensen J.; Mroczkowski B.; Bender S.; Kania R. S.; Edwards M. P. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [DOI] [PubMed] [Google Scholar]
- Human Kinome. http://kinase.com/human/kinome/ (accessed June 2013).
- DDR1 had a POC of 18% in the presence of 1 μM compound 4.
- The panel of GPCRs includes receptors 5HT1A, ADRA1A, ADORA2, CB1, CCR2, CXCR2, D2, H1, M1, MOR, NK1, SST4, and ADRB1.
- At 100 mg/kg b.i.d. dosing, significant animal health concerns (cold, lethargic, hunched, dehydrated, and diarrhea) were noted at day 8, prior to sacrifice.
- Lau T.; Chan E.; Callow M.; Waaler J.; Boggs J.; Blake R. A.; Magnuson S.; Sambrone A.; Schutten M.; Firestein R.; Machon O.; Korinek V.; Choo E.; Diaz D.; Merchant M.; Polakis P.; Holsworth D. D.; Krauss S.; Costa M. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–3144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.