

Abstract

We report the development of a new trifluoromethyltriazolobenzoxazepine series of squalene synthase inhibitors. Structure–activity studies and pharmacokinetics optimization on this series led to the identification of compound 23 (DF-461), which exhibited potent squalene synthase inhibitory activity, high hepatic selectivity, excellent rat hepatic cholesterol synthesis inhibitory activity, and plasma lipid lowering efficacy in nonrodent repeated dose studies.
Keywords: Squalene synthase, inhibitor, dyslipidemia, triazolobenzoxazepine
In recent years, the number of arteriosclerosis, ischemic heart diseases, and ischemic brain diseases attributed to arteriosclerosis is increasing around the world due to the spread of the Western dietary pattern and the growing number of elderly affected people. Hypercholesterolemia is one of the primary risk factors of arteriosclerosis. To treat the disease, it is effective to administer medicines for reducing the serum low density lipoprotein (LDL) cholesterol level. The cardiovascular risk reduction of hydroxymethylglutraryl-CoA (HMG-CoA) reductase inhibitors (statins) is principally attributed to their LDL lowering effects as demonstrated in numerous randomized clinical trials.1−3 However, statins have potential adverse effects, such as myotoxicity, muscle pain, and, in very rare cases, rhabdomyolysis4,5 because the inhibition of HMG-CoA reductase also interferes with the synthesis of many nonsteroidal isoprenoid molecules. Cerivastatin, one of the second generation statins, was withdrawn from the world market in 2001, due to its adverse effects.6 Thus, it is preferable to prevent the cholesterol biosynthesis by targeting an enzyme, which lies downstream of farnesyl pyrophosphate in the cholesterol biosynthesis pathway, without interruption to the biosynthesis of essential physiological components, such as ubiquinone and dolichol.
Squalene synthase is an attractive target mainly because it is the first enzyme involved in the sterol biosynthesis.7,8 The inhibitors of the enzyme do not inhibit nonsteroidal biomolecules.9−12 Although there are already known inhibitors of squalene synthase, it is still difficult to confirm that these compounds have the required sufficient cholesterol lowering potential.7,8,13−18
Our research group has already reported the lead identifications and initial optimizations of two series of small molecule squalene synthase inhibitors; one is an open form benzhydrol series, another is a tricyclic pyrrolobenzoxazepine series, demonstrating plasma lipid-lowering efficacies in preclinical animal models.
Exploratory medicinal chemistry efforts focused on initial lead 1.19 The highly potent alkoxy-aminobenzhydrol compound 2 was obtained,20 through incorporations of both the nipecotic acid part at the end of the side chain and the bulky alkoxy part at the aniline ring’s ortho position; the benzhydrol compounds formed unique 11-membered ring active conformations with intramolecular hydrogen bonds, between the benzhydrol hydroxyl part and the side chain amide carbonyl oxygen, in the squalene synthase catalytic domain (Figure 1).
Figure 1.

Structures of 1–3 and TAK-475.
Although their IC50 values reached a single-digit nanomolar order, the in vivo efficacies were not enough for acquiring the clinical candidate. An undesirable feature of the benzhydrol series might be an inadequate stability of the hydroxyl groups, especially in acidic conditions.
In order to achieve a more potent in vivo effective compound, a new tricyclic scaffold was designed, mainly be cause the oxazepine ring’s ether bond was more stable than the intramolecular hydrogen bond. Consequently, an improved series of squalene synthase inhibitors, based on the pyrrolobenzoxazepine template, had been identified.21 Successively, we focused our efforts on the optimization of its side chain to advance the tricyclic series. As a result, highly potent compound 3 was garnered, which demonstrated excellent plasma lipid-lowering effects in marmoset oral repeated dose studies.
Subsequently, two concerns of the template were at the forefront; one was its chemical stability, and the other was its organ selectivity after administration. First, the template had a slightly reactive carbon atom in its pyrrole part, which probably increased the possibility of some adverse effects by reacting with the in vivo substrate. To ensure the safety of long-term administration as lipid-lowering medicine, we endeavored to solve this issue. Second, compound 3 showed no organ selectivity, where there was room for improvement of its hepatic selectivity which was a crucial issue as a squalene synthase inhibitor, whose target organ is the liver. Recently, highly hepatic selectivity was demanded for inhibiting the cholesterol biosynthetic cascade, even with an HMG-CoA reductase inhibitor,22 because some cholesterol derived biomolecules have essential roles in peripheral organs. Therefore, disrupting the cholesterol biosynthesis in other organs might increase the risk for adverse effects.
To minimize the possibility of unforeseen adverse effects, our research effort focused on the optimization of the tricyclic template. Exchanging the pyrrole part in the tricyclic template to other heterocycles was attempted to avoid the reactive carbon atom in the ring, at the same time, decreasing the lipophilicity, which probably increased the liver selectivity. From past research experience, our inhibitors might be recognized and selectively taken into the hepatic cell by organic anion transporters, expressed on a hepatic cell surface, in addition to the passive diffusion. We anticipated that decreasing the compound’s lipophilicity suppressed the diffusional cell permeability without significant effect on the active transportation, moreover improving the pharmacokinetic profile.
We designed and prepared new tricyclic templates, which incorporated more nitrogen containing five-membered heteroaryl rings instead of pyrrole rings, such as pyrazole and triazole rings. In our preliminary research, we found substituted triazole ring attached derivatives had comparable squalene synthase inhibitory (SSI) activity23 with pyrrolobenzoxazepine (Table S1, Supporting Information).
Furthermore, the optimization of triazole’s substituent was surveyed tentatively on pyrazole 4-carboxylic acid derivatives, which were appropriate for synthesis (Table 1). To obtain the profile of the derivatives, their SSI activity, cholesterol synthesis inhibitory (CSI) activity in rat hepatic cells,23 and in vivo rat hepatic cholesterol synthesis inhibitory (in vivo CSI) activity were evaluated with lipophilicity and permeability.
Table 1. Modification of the Triazole Ring Substituent in trans-Racematea.
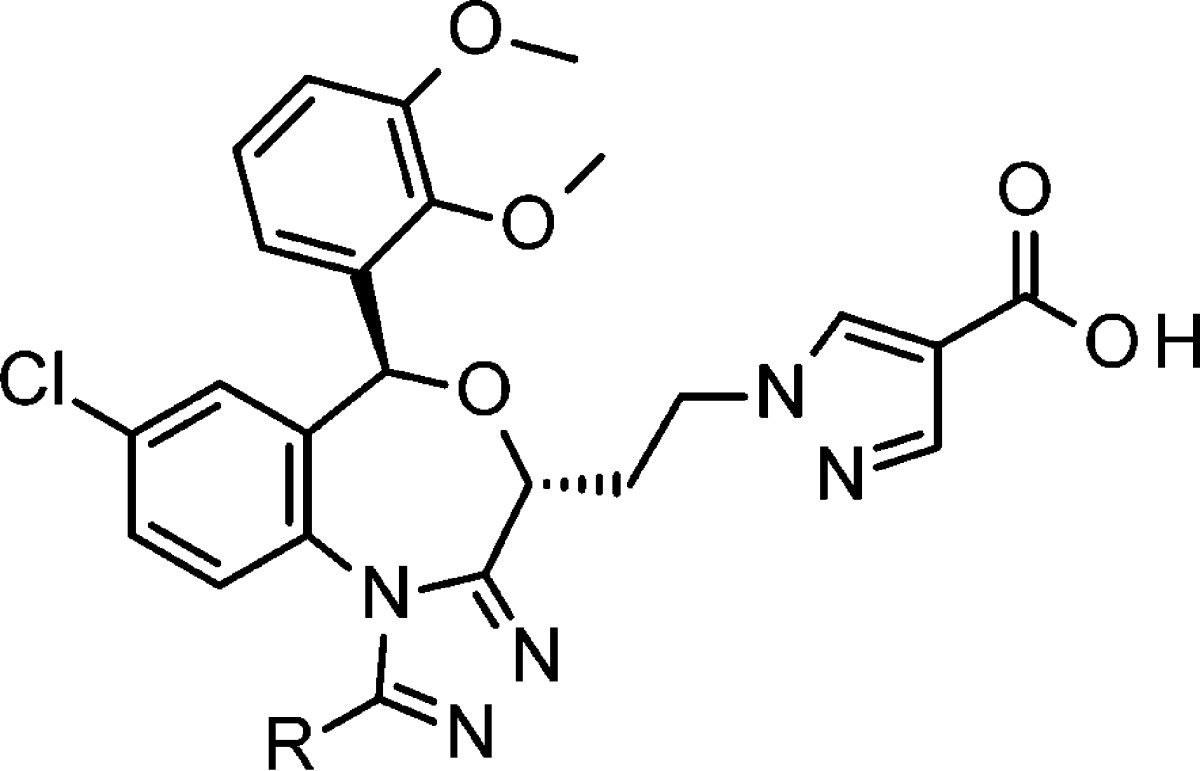
| compd | R | SSI (IC50 nM) | CSI (IC50 nM) | logD6.4 | PAMPA (pH 7.4) | in vivo CSI (3 mg/kg %) |
|---|---|---|---|---|---|---|
| 4 | Me | 36 | 290 | –0.4 | <2 | 30 |
| 5 | iPr | 11 | 91 | 0.37 | 4.7 | 15 |
| 6 | CH2F | 6.0 | 77 | 48 | ||
| 7 | CF3 | 3.2 | 130 | 0.83 | 10 | 71 |
SSI: squalene synthase inhibitory activity. CSI: cholesterol synthesis inhibitory activity in rat hepatic cells. PAMPA: cell permeability score at pH 7.4. In vivo CSI: hepatic cholesterol inhibition (%) in rats at 1 h after 3 mg/kg single dose oral administration. No data = not tested.
At first, methyl-substituted compound 4 was prepared and evaluated. While 4 had middle SSI activity, the permeability was unmeasurable due to its very low lipophilicity (logD7.4 = −0.4). To add more lipophilicity, we prepared larger isopropyl derivative 5, which showed slightly higher permeability with the increasing lipophilicity (PAMPA = 4.7, logD7.4 = 0.37). Although 5 showed improved SSI and CSI activity (IC50 = 11 nM and 91 nM, respectively), the in vivo efficacy declined (in vivo CSI = 15% inhibition); probably due to the metabolic instability.
To obtain further improved cell permeability, we tried to reduce triazole’s relatively high electron density by introducing electron-deficient fluorine atoms to the methyl group, attached to the ring. As predicted, monofluoromethyl compound 6 showed improved in vivo efficacy (in vivo CSI = 48% inhibition). Moreover, further electron-deficient trifluoromethyl compound 8 exhibited higher SSI activity, good permeability, and proceeded in vivo efficacy (SSI = 3.2 nM, PAMPA = 10, and in vivo CSI = 71%). We solved the permeability issue of the triazole containing the new template.
To access the series of compounds with heteroaryl containing a tricyclic template, the amino part of 8 was protected with the 2,4-dimethoxybenzyl group, then reacted with acid chloride to generate fumaric amide 10. Subsequent Michael cyclization gave benzoxazepine 11. Thioamide 13 was prepared by radical deprotection with ammonium cerium(IV) nitrate (CAN) and thioamidation with Lawesson’s reagent. Following sequential reactions with hydrazine, acid chloride, and sequential acidic cyclization gave triazole containing the tricyclic template. Subsequent ester part reduction gave 15–18 following methanesulfonylation, and pyrazole part substitution gave ester pyrazole compounds, which were hydrolyzed to the final pyrazolecarboxylic acids 4–7. Compounds 19–23 were prepared by optical resolution of 14 and hydrolysis (Scheme 1).
Scheme 1.

To further improve the pharmacokinetic profile of our compound, we focused on decreasing the molecular weight mainly because smaller compounds would have better hepatic selectivity due to the lower peripheral diffusion. After considerable effort, we revealed that only the acetic acid side chain attached derivatives have excellent potency, higher SSI and CSI activity, and remarkably improved in vivo persistent efficacy, one of the important factors as lipid lowering medicine.10 As shown in Table 2, simple 2,3-dimethoxyphenyl 19 demonstrated improved and persistent in vivo efficacy in the rat time course hepatic cholesterol synthesis inhibitory study (1 mg/kg, po single dose: 1 h 88%, 4 h 78%, and 7 h 45% inhibition).
Table 2. Optimization of the Upper Phenyl Ring Part in (4R, 6S)-Isomera,b.
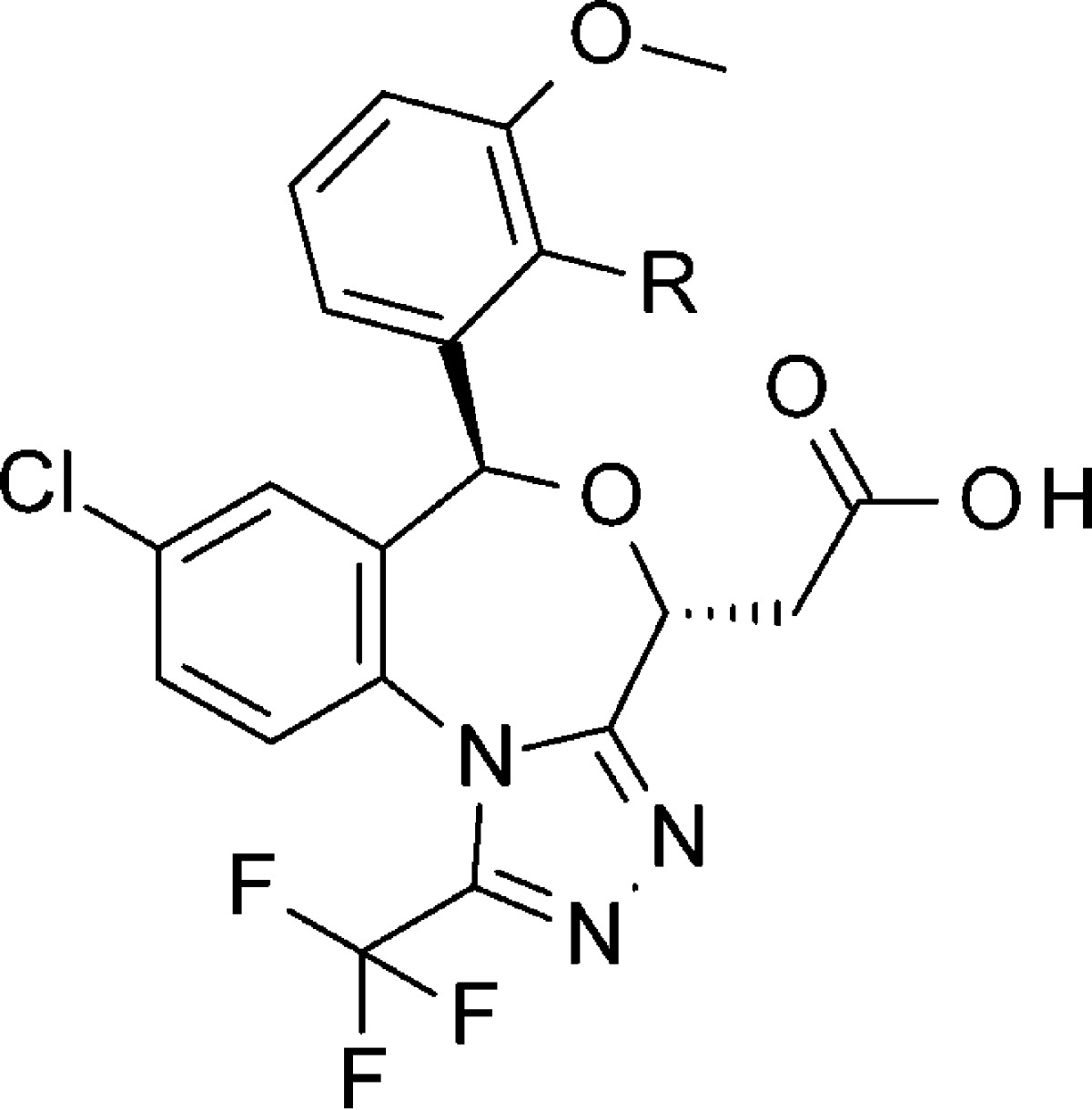
| in vivo CSI (1 mg/kg, % inhibition) |
||||||
|---|---|---|---|---|---|---|
| compd | R | SSI (IC50 nM) | CSI (IC50 nM) | 1 h | 4 h | 7 h |
| 19 | OMe | 1.6 | 38 | 88 | 78 | 45 |
| 20 | OCF3 | 0.7 | 28 | 77 | 67 | 48 |
| 21 | Et | 0.36 | 18 | 76 | 57 | 21 |
| 22 | Me | 0.85 | 46 | 87 | 69 | 45 |
| 23 | Cl | 1.1 | 19 | 93 | 77 | 55 |
The same configuration as TAK-475.
SSI: squalene synthase inhibitory activity. CSI: cholesterol synthesis inhibitory activity in rat hepatic cell. In vivo CSI: hepatic cholesterol inhibition (%) in rat at 1, 4, and 7 h after 1 mg/kg single dose oral administration.
At the next stage, we aimed to adjust the balance between the in vivo efficacy and liver selectivity by optimization of the remaining common part that was identified to be suitable for physical property fine-tuning: the upper benzene ring substituent. Focusing on 2-position substituent of the phenyl ring, we prepared 2-trifluoromethoxy, methyl, ethyl, and chloro derivatives 20–23. 2-Trifluoromethoxy-3-methoxyphenyl 20 and 2-ethyl-3-methoxyphenyl 21 showed subnanomolar SSI activity (the IC50 values were 0.7 and 0.36 nM, respectively); however, their in vivo efficacy and durations were slightly decreased. 2-Methyl-3-methoxyphenyl 22 showed strong in vivo CSI potency, the same as 19. Especially, 2-chloro-3-methoxyphenyl 23 demonstrated the most potent in vivo CSI efficacy (1 mg/kg po, 1 h 93%, 4 h 77%, and 7 h 55%).
Successively, we investigated the plasma lipid lowering study in marmoset with selected potential compounds 19, 22, 23, and Takeda’s predecessor clinical compound TAK-475 as positive control, at 30 mg/kg/day repeated oral administrations for 7 days. As shown in Table 3, all our compounds showed significant reduction of serum non-HDL cholesterol levels from 42% to 53% reduction, especially 23 demonstrated the highest 53% reduction; additionally, plasma triglyceride (TG) levels reducing potency were observed from 24% to 34% reduction, and at the same time, TAK-475 reduced 19% on non-HDL and 25% on TG).
Table 3. Plasma Lipid Lowering Effects in Marmoset 30 mg/kg/day Orally Repeated Doses for 7 Daysa.
| compd | TC (%) | non-HDL C (%) | HDL C (%) | TG (%) |
|---|---|---|---|---|
| TAK-475 | 17 | 19 | 10 | 25 |
| 19 | 29 | 44 | 3 | 29 |
| 22 | 32*** | 42*** | 8 | 34*** |
| 23 | 39*** | 53*** | 16 | 24** |
TC: total cholesterol. non-HDL C: non-HDL cholesterol. HDL C: HDL cholesterol. TG: triglyceride. Data are shown as the mean values of percentage changes from initial values (n = 7, 8). **p < 0.01 and ***p < 0.001 vs. initial values.
Next, selected compounds’ liver selectivity was measured in rats at 3 mg/kg orally single dose administration. 2-Chloro compound 23 demonstrated the highest hepatic concentration and better selectivity (liver concentration 2894 ng/g and Kpliver 80 at 1 h) compared to 19 (liver conc. 1045 ng/g and Kpliver 66 at 1 h), and 2-methyl compound 22 showed improper selectivity (Table S2, Supporting Information).
On the basis of favorable evaluation results, we chose 23 as a candidate to evaluate the higher characterization study, which showed the highest plasma lipid lowering potency in marmoset, and the highest liver selectivity and persistent in vivo CSI efficacy in rats.
In the further biological activity characterization, 23 indicated excellent ED50 value in rats (0.11 mg/kg), over 30 times stronger than TAK-475 (3.8 mg/kg). Finally, the in vivo CSI potential comparison study was evaluated with atorvastatin, HMG-CoA reductase inhibitor in rats (Figure 2). Consequently, 23 exhibited admirable, cholesterol synthesis inhibitory potential, which was better than atorvastatin at the same dose.
Figure 2.
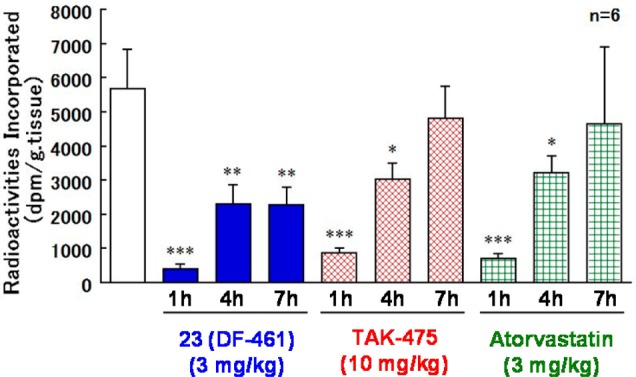
Evaluation of cholesterol synthesis inhibitory potency in rat with 14C-acetic acid.
In summary, a novel series of squalene synthase inhibitors were discovered by exchanging the pyrrole part to the trifluoromethyltriazole part in the tricyclic template of lead compound 3. Further optimization of the new template has resulted in the identification of acetic acid side chain derivatives with improved plasma lipid lowering potency in preclinical species. On the basis of the most potent in vivo efficacy, liver selectivity, and other desirable profiles, the arginine salt of 23 (DF-461) has been chosen as the drug development candidate.
Acknowledgments
We thank the members of Cardiovascular-Metabolics Research Laboratories for the biological assays and Daiichi Sankyo RD NOVARE Co., Ltd., for the elemental analysis. Also, we would like to acknowledge Dr. Hisashi Takahashi for his helpful support in the preparation of this manuscript.
Supporting Information Available
Additional tables (S1 and S2), synthetic procedures and characterization data for compounds, and biological evaluation procedure. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Present Address
† Faculty of Pharmaceutical Science, Hoshi University, Tokyo 142-0063, Japan.
Supplementary Material
References
- Tobert J. A. Losvastatin and beyond: the history of the HMG-CoA reductase inhibitor. Nat. Rev. Drug. Discovery 2003, 2, 517–526. [DOI] [PubMed] [Google Scholar]
- Havel R. J.; Rapaport E. Management of primary hyperlipidemia. New Eng. J. Med. 1995, 332, 1491–1498. [DOI] [PubMed] [Google Scholar]
- Shepherd J.; Cobbe S. M.; Ford I.; Isles C. G.; Lorimer A. R.; Macfarlane P. W.; McKillop J. H.; Packard C. J. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N. Engl. J. Med. 1995, 333, 1301–1308. [DOI] [PubMed] [Google Scholar]
- Hodel C. Myopathy and rhabdomyolysis with lipid-lowering drugs. Toxicol. Lett. 2002, 128, 159–168. [DOI] [PubMed] [Google Scholar]
- Thompson P. D.; Clarkson P.; Karas R. H. Statin-associated myopathy. JAMA. 2003, 289, 1681–1690. [DOI] [PubMed] [Google Scholar]
- Furberg C. D.; Pitt B. Withdrawal of cerivastatin from the world market. Curr. Control Trials Cardiovasc. Med. 2001, 2, 205–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T.; Kakuta H.; Moritani H.; Ugawa T.; Yanagisawa I. Synthesis and biological evaluation of novel propylamine derivatives as orally active squalene synthase inhibitors. Bioorg. Med. Chem. 2004, 12, 5899–5908. [DOI] [PubMed] [Google Scholar]
- Miki T.; Kori M.; Mabuchi H.; Tozawa R.; Nishimoto T.; Sugiyama Y.; Teshima K.; Yukimasa H. Synthesis of novel 4,1-benzoxazepine derivatives as squalene synthase inhibitors and their inhibition of cholesterol synthesis. J. Med. Chem. 2002, 45, 4571–4580. [DOI] [PubMed] [Google Scholar]
- Biller S. A.; Neuenschwander K.; Ponpipom M. M.; Poulter C. D. Squalene synthase inhibitors. Curr. Pharm. Des. 1996, 2, 1–40. [Google Scholar]
- Gonzalez-Pacanowska D.; Arison B.; Havel C. M.; Watson J. A. Isopentenoid synthesis in isolated embryonic Drosophila cells. Farnesol catabolism and ω-oxidation. J. Biol. Chem. 1988, 263, 1301–1306. [PubMed] [Google Scholar]
- Bergstrom J. D.; Kurtz M. M.; Rew D. J.; Amend A. M.; Karakas J. D.; Bostedor R. G.; Bansal V. S.; Dufresne C.; VanMiddlesworth F. L.; Hensens O. D. Zaragozic acids: a family of fungal metabolites that are picomolar competitive inhibitors of squalene synthase. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostedor R. G.; Karkas J. D.; Arison B. H.; Bansal V. S.; Vaidya S.; Germershausen J. I.; Kurtz M. M.; Bergstrom J. D. Farnesol-derived dicarboxylic acids in the urine of animals treated with zaragozic acid A or with farnesol. J. Biol. Chem. 1997, 272, 9197–9203. [DOI] [PubMed] [Google Scholar]
- Griebenow N.; Flessner T.; Buchmueller A.; Raabe M.; Bischoff H.; Kolkhof P. Identification and optimization of tetrahydro-2H-3-benzazepin-2-ones as squalene synthase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2554–2558. [DOI] [PubMed] [Google Scholar]
- Griebenow N.; Buchmueller A.; Kolkhof P.; Schamberger J.; Bischoff H. Identification of 4H,6H-[2]benzoxepino[4,5-c][1,2]oxazoles as novel squalene synthase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 3648–3653. [DOI] [PubMed] [Google Scholar]
- Hiyoshi H.; Yanagimachi M.; Ito M.; Saeki T.; Yoshida I.; Okada T.; Ikuta H.; Shinmyo D.; Tanaka K.; Kurusu N.; Tanaka H. Squalene synthase inhibitors reduce plasma triglyceride through a low-density lipoprotein receptor-independent mechanism. Eur. J. Pharmacol. 2001, 431, 345–352. [DOI] [PubMed] [Google Scholar]
- Brown G. R.; Clarke D. S.; Foubister A. J.; Freeman S.; Harrison P. J.; Johnson M. C.; Mallion K. C.; McCormick J.; McTaggart F.; Reid A. C.; Smith G. J.; Taylor M. J. Synthesis and activity of a novel series of 3-biarylquinuclidine squalene synthase inhibitors. J. Med. Chem. 1996, 39, 2971–2979. [DOI] [PubMed] [Google Scholar]
- Usui H.; Kagechika K.; Nagashima H.. WO1998/029380.
- Usui H.; Katakura S.; Suzuki M. Jpn. Kokai Tokkyo Koho. 2001, 131.p (JP 2001/354587).. [Google Scholar]
- Ichikawa M.; Yokomizo A.; Itoh M.; Sugita K.; Usui H.; Shimizu H.; Suzuki M.; Terayama K.; Kanda A. Discovery of a new 2-aminobenzhydrol template for highly potent squalene synthase inhibitors. Bioorg. Med. Chem. 2011, 19, 1930–1949. [DOI] [PubMed] [Google Scholar]
- Ichikawa M.; Yokomizo A.; Itoh M.; Haginoya N.; Sugita K.; Usui H.; Terayama K.; Kanda A. Discovery of atrop fixed alkoxy-aminobenzhydrol derivatives: Novel, highly potent and orally efficacious squalene synthase inhibitors. Bioorg. Med. Chem. 2011, 19, 5207–5224. [DOI] [PubMed] [Google Scholar]
- Ichikawa M.; Ohtsuka M.; Ohki H.; Ota M.; Haginoya N.; Itoh M.; Sugita K.; Usui H.; Suzuki M.; Terayama K.; Kanda A. Discovery of novel tricyclic compounds as squalene synthase inhibitors. Bioorg. Med. Chem. 2012, 20, 3072–3093. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn J. A.; Litchfield J.; Hutchings R.; Cheng X. M.; Larsen S. D.; Auerbach B.; Bush M. R.; Lee C.; Erasga N.; Bowles D. M.; Boyles D. C.; Lu G.; Sekerke C.; Askew V.; Hanselman J. C.; Dillon L.; Lin Z.; Robertson A.; Olsen K.; Boustany C.; Atkinson K.; Goosen T. C.; Sahasrabudhe V.; Chupka J.; Duignan D. B.; Feng B.; Scialis R.; Kimoto E.; Bi Y. A.; Lai Y.; El-Kattan A.; Bakker-Arkema R.; Barclay P.; Kindt E.; Le V.; Mandema J. W.; Milad M.; Tait B. D.; Kennedy R.; Trivedi B. K.; Kowala M. Discovery of novel hepatoselective HMG-CoA reductase inhibitors for treating hypercholesterolemia: A bench-to-bedside case study on tissue selective drug distribution. Bioorg. Med. Chem. Lett. 2011, 21, 2725–2731. [DOI] [PubMed] [Google Scholar]
- Shechter I.; Klinger E.; Rucker M. L.; Engstrom R. G.; Spirito J. A.; Islam M. A.; Boettcher B. R.; Weinstein D. B. Solubilization, purification, and characterization of a truncated form of rat hepatic squalene synthetase. J. Biol. Chem. 1992, 267, 8628–8635. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.