

Abstract

11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) catalyzes the conversion of inactive glucocorticoid cortisone to its active form, cortisol. The glucocorticoid receptor (GR) signaling pathway has been linked to the pathophysiology of diabetes and metabolic syndrome. Herein, the structure–activity relationship of a series of piperazine sulfonamide-based 11β-HSD1 inhibitors is described. (R)-3,3,3-Trifluoro-2-(5-(((R)-4-(4-fluoro-2-(trifluoromethyl)phenyl)-2-methylpiperazin-1-yl)sulfonyl)thiophen-2-yl)-2-hydroxypropanamide 18a (HSD-621) was identified as a potent and selective 11β-HSD1 inhibitor and was ultimately selected as a clinical development candidate. HSD-621 has an attractive overall pharmaceutical profile and demonstrates good oral bioavailability in mouse, rat, and dog. When orally dosed in C57/BL6 diet-induced obesity (DIO) mice, HSD-621 was efficacious and showed a significant reduction in both fed and fasting glucose and insulin levels. Furthermore, HSD-621 was well tolerated in drug safety assessment studies.
Keywords: 11β-hydroxysteroid dehydrogenase type I (HSD1), diet-induced obesity, type II diabetes, piperazine sufonamides
Glucocorticoid hormones are key regulators of a wide range of biological processes such as the control of immune and stress responses as well as modulation of energy metabolism. Glucocorticoids (cortisol in humans and corticosterone in mice and rats) stimulate hepatic glucose production and suppress insulin-mediated glucose uptake in peripheral tissues (i.e., adipose and muscle). 11β-Hydroxysteroid dehydrogenase type I (11β-HSD1),1−1g a reduced β-nicotinamide adenine dinucleotide phosphate (NADPH)-dependent enzyme, predominantly acts as a reductase in vivo, converting inactive, nonreceptor binding cortisone to active, receptor-binding cortisol in tissues such as liver, adipose, vasculature, brain, and macrophages.2 The enzyme is a tetramer consisting of two dimers each with an independent active site.2 It has been proposed that 11β-HSD1 reductase activity exists predominantly in metabolic tissues because of the increased ratio of NADPH to β-nicotinamide adenine dinucleotide phosphate (NADP) within the endoplasmic reticulum (ER) lumen and/or through direct interactions with NADPH-generating hexose-6-phosphate dehydrogenase (H6PDH), an enzyme that has been associated with cortisone reductase deficiency in humans.3 Cortisone itself is generated by the action of 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) with cortisol using NADP as a cofactor. 11β-HSD1 levels are highest in liver and adipose tissues as well as in the central nervous system, whereas 11β-HSD2 is mainly expressed in the kidney and colon.4
The rationale for 11β-HSD1 as a therapeutic target for the treatment of diabetes and the metabolic syndrome is based on data from tissue-specific overexpression in transgenic mice5,5b and mice 11β-HSD knockout experiments.6,6b In addition, 11β-HSD1 activity in adipose tissue also correlates positively with body mass index (BMI), fat percentage, and fasting glucose and insulin levels in humans. Recent studies have demonstrated that whole-body 11β-HSD1 activity is elevated in obese men with type 2 diabetes, whereas liver 11β-HSD1 activity is relatively unchanged, suggesting that disease suppression via 11β-HSD1 inhibition is likely to be more effective in obese patients with type 2 diabetes.7
Several classes of potent and selective 11β-HSD1 inhibitors have been reported in the literature.8−8k Some inhibitors, including our own,8,8b have displayed in vivo efficacy in animal models related to diabetes.9−9e Data from phase II clinical trials for 11β-HSD1 inhibitor INCB-173910 showed improved hepatic and peripheral insulin sensitivity and a reduction in fasting plasma glucose and cholesterol observed after 28 days of treatment in type II diabetic patients. Decreased triglyceride levels and improvement in blood pressure were also reported.
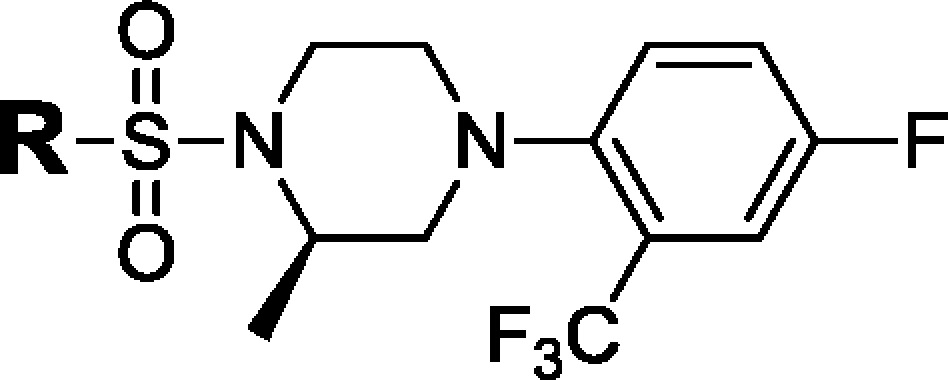
We previously disclosed a series of compounds exemplified by 1 and 2, which displayed potent and selective inhibition against both human and rodent 11β-HSD1 in vitro. The compounds also showed oral efficacy in the cortisone-induced hyperinsulinemia rat model8 and in the diet-induced obesity (DIO) mouse model8b (Figure 1). In the continuation of our 11β-HSD1 program, we are reporting on the discovery of our clinical development candidate 18a [HSD-621, (R)-3,3,3-trifluoro-2-(5-((R)-4-(4-fluoro-2-(trifluoromethyl)-phenyl)-2-methylpiperazin-1-ylsulfonyl)thiophen-2-yl)-2-hydroxypropanamide]. Selected characterization and preclinical data for HSD-621 will be disclosed.
Figure 1.

Potent and selective piperazine sulfonamides as 11β-HSD1 inhibitors.
On the basis of the profile of our earlier compounds,8,8b we imposed additional requirements when selecting compounds to move forward in our screening cascade. Specifically, we were interested in compounds with the following profile: (i) potent cellular activity, (ii) good microsomal stability, and (iii) potent adipose ex vivo activity. Among these considerations, adipose ex vivo activity became the most significant driver in terms of compound selection for in vivo efficacy studies. This stemmed from the earlier observation of poor in vivo results from compounds possessing high liver ex vivo activity alone.8b This finding is in line with the report that liver-specific overexpression of 11β-HSD1 in transgenic mice causes insulin resistance without obesity,11 possibly indicating the importance of targeting 11β-HSD1 outside of the liver, particularly in the adipose.7
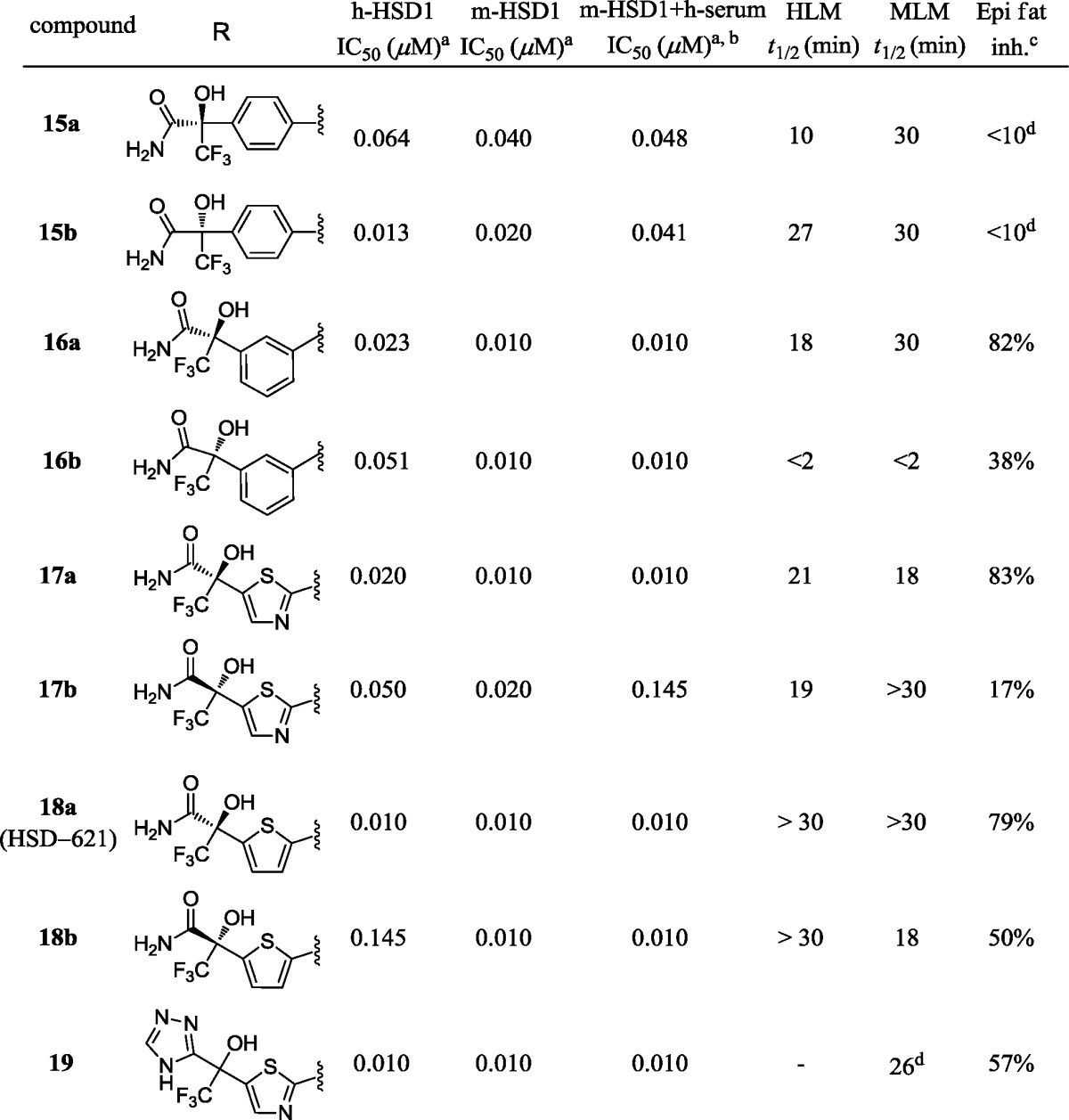
Our earlier work taught us that the active site of 11β-HSD1 provides opportunities for both electrostatic and van der Waals interactions (e.g., 1(8a) and 2(8b)). Among these interactions was a hydrogen bond between one of the sulfonamide oxygens with the backbone nitrogen of A172 (Figure 2). This was conserved among all compounds in our series. The overall binding site can be described as two pockets, one on either side of this conserved hydrogen bond. Pocket A contains the active site of the enzyme as well as NADP. It is a lipophilic region formed by the side chains of residues I121, T124, L126, V180, and Y183. Some inhibitors were shown to reach outside the hydrophobic pocket and interact with the cofactor NADP. The second side of the binding pocket (pocket B) holds the remainder of the steroid substrate and is formed, in part, by the dimer partner of the enzyme monomer to which the inhibitor is bound. Key residues in this pocket include Y177 and P178.
Figure 2.
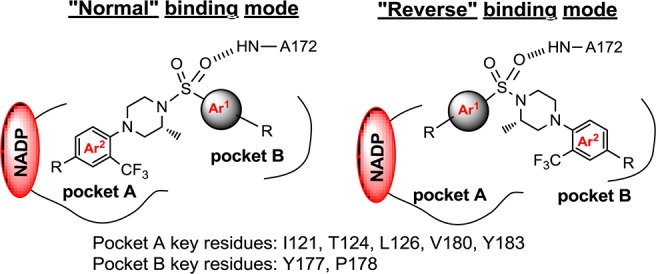
“Normal” and “reverse” binding modes of piperazine sulfonamides as 11β-HSD1 inhibitors in human 11β-HSD1 enzyme.
We observed two binding modes with our 11β-HSD1 inhibitors: the “normal” binding mode and the “reversed” binding mode, both of which have a conserved hydrogen bond between one of the sulfonamide oxygens and the NH of A172 (Figure 2). In the “normal” binding mode, the trifluoromethyl group on the phenyl ring (Ar2) of 1 and 2, common to all analogues in the piperazine phenyl sulfonamide series, sits in the lipophilic pocket A. In the “reverse” binding mode, the inhibitors rotate 180° centered at the conserved hydrogen bond and the phenyl ring Ar2 now occupies pocket B instead of pocket A.
In the piperazine phenyl sulfonamide series, we had observed that meta- versus para-substitution on the phenyl ring Ar1 of 1 had an effect on potency as well as ex vivo activity (Figure 1).8a We reasoned that a five-membered ring might offer an intermediate positioning of substituents that the six-membered phenyl sulfonamide scaffold could not provide. Guided by structural information, a number of potent and selective piperazine thiophene sulfonamides were designed and synthesized (Scheme 1 and Table 1).
Scheme 1.

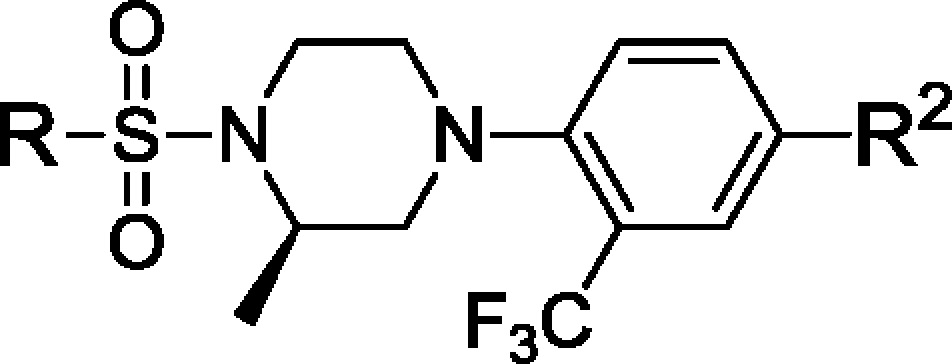
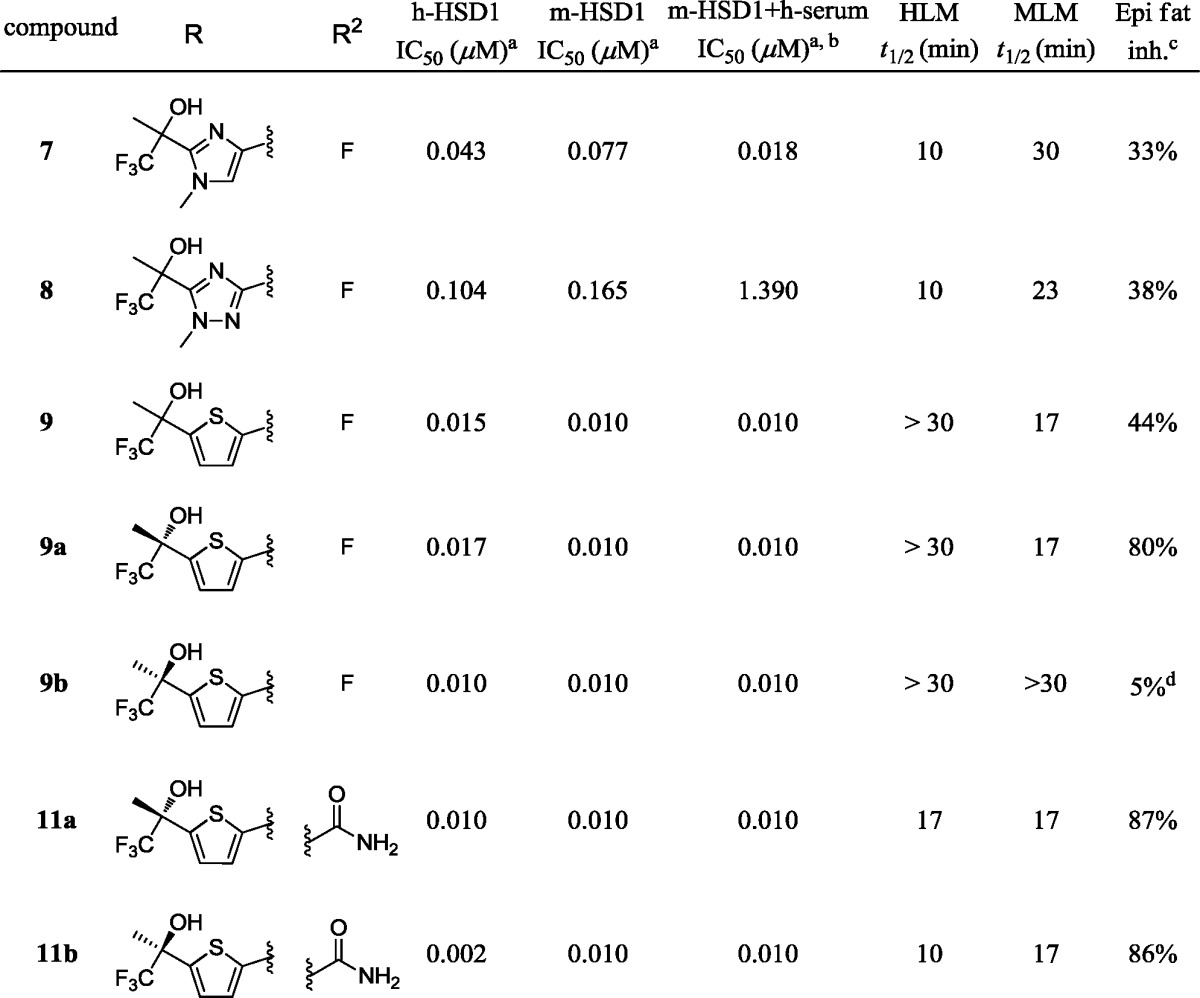
Table 1. Inhibitory Activity, Microsomal Stability, and Epididymal Fat ex Vivo Inhibition of Selected Compounds.
The assay was performed in duplicate; the compounds were tested in 11-point 2-fold dilutions, and the standard error of IC50 was <10%; the lower threshold of this assay was 10 nM.
10% (v/v) human serum was added to the cell culture media.
po dosed at 10 mg/kg; results obtained 5 h after dosing.
Not significant.
We began by replacing the left-hand side phenyl group Ar1 in compounds 1 and 2 with five-membered rings as exemplified by imidazole and triazole analogues 7 and 8 (Table 1 and Scheme 1). These compounds bear a lipophilic trifluoromethyl propan-2-ol moiety, an optimally refined group in our first clinical compound HSD-016.12 Both compounds were easily synthesized by halogen/lithium exchange at the C2 positions followed by quenching by trifluoroacetone. Encouragingly, both compounds were found to be active against both human and mouse 11β-HSD1 enzymes with moderate fat ex vivo activity of 33 and 38%, respectively, 5 h after oral dosing at 5 mg/kg.
Continuing efforts led to the discovery of the thiophene analogue 9 (Table 1). In our cell-based assays, 9 was very potent against human and mouse 11β-HSD1 enzymes (15 and 10 nM, respectively) and displayed reasonable stability against both human liver microsomes (HLM, >30 min) and mouse liver microsomes (MLM, 17 min). More encouragingly, a 44% inhibition of 11β-HSD1 in epididymal fat (epi fat) was observed in the ex vivo assay. Interestingly, single diasteromers 9a and 9b had distinct biological profiles, in which the S-isomer 9a demonstrated much higher inhibition than the R-isomer 9b (80 vs 5%, Table 1).
It is known that engaging the cofactor NADP is capable of modulating the binding efficiency.13 Molecular modeling suggested that a favorable hydrogen bond could exist between a carboxamide group off the phenyl ring (Ar2) and the phosphate backbone of the NADP in the “normal” bind mode, and this was borne out subsequently in crystal structures of related compounds (data not shown). Thus, the fluoro group at the phenyl C4 position in compound 9 was replaced by a carboxamide group to give 11a and 11b after chiral separation (Scheme 1 and Table 1). While no measurable improvement in cell-based potency was observed due to the 10 nM assay threshold, potent fat ex vivo inhibition was observed for both compounds 11a and 11b (87 and 86%, respectively). Unfortunately, compounds 11a and 11b have relatively short half-lives in both human and mouse microsomes (17 min/17 and 10 min/17 min, respectively). Additionally, both compounds displayed inhibition against the cytochrome P450 isozyme 2C9 with IC50 values in the 100 nM range. In comparison, compounds 9 and 9a were not substrates of P450 enzymes.
Although compounds 11a and 11b failed to meet our criteria for further advancement, they prompted us to further explore alternative electrostatic interactions with NADP through the “reverse” binding mode. In addition to maintaining the key hydrogen bond at the center of the molecule, we incorporated functionalities capable of forming hydrogen bond interactions with NADP with a donor group on the aryl sulfonamide part of the molecule while keeping constant the 2-trifluoromethyl-4-fluorophenyl group to maintain the lipophilic interactions with pocket B (Figure 2).
We began this effort by replacing the methyl group of the trifluoromethylpropan-2-ol moiety in 9 with a carboxamide group in the phenyl sulfonamide moiety, leading to the para- and meta-substituted phenyl analogues 15a,b and 16a,b (Scheme 2 and Table 2). This substitution was predicted by molecular modeling to yield compounds with hydrogen bond interactions between the carboxamide moiety and the cofactor NADP in the “reverse” binding mode. Increased microsomal stability was also predicted due to the overall increase in the polarity of the compounds. In addition, we also noticed that the dehydration of tertiary alcohol in 9a occurs under acid conditions. Removal of the α-protons with a C(O)NH2 group was anticipated to mitigate the problem.
Scheme 2.
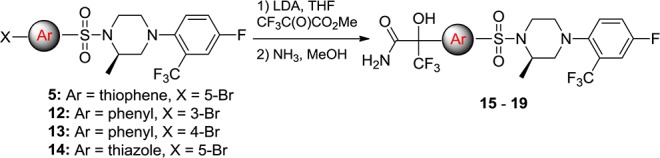
Table 2. Inhibitory Activity, Microsomal Stability, and Epididymal Fat ex Vivo Inhibition of Compounds Targeting the “Reverse” Binding Mode.
The assay was performed in duplicate; the compounds were tested in 11-point 2-fold dilutions, and the standard error of IC50 was <10%; the lower threshold of this assay was 10 nM.
10% (v/v) human serum was added to the cell culture media.
po at 10 mg/kg otherwise noted, results obtained 5 h after dosing.
Treatment of 12 and 13 with n-BuLi followed by the addition of methyl 4,4,4-trifluoro-2,3-dioxobutanoate gave the desired ester intermediate, which was then amidated with concentrated NH3 to give compounds 15a,b and 16a,b, respectively (Scheme 2). The difference in ex vivo efficacy for these two substitution patterns was found to be consistent with our previous observations, the meta-substitution being favored8a as shown in compounds 16a and 16b (Table 2). The R-isomer 16a showed more robust ex vivo inhibition (82 vs 38%) as well as increased human and mouse microsomal stability (18 and 30 min vs 2 and 2 min, respectively).
The preference for the meta-substitution prompted us to revisit the five-membered ring heterocycles as exemplified in the thiazole and thiophene analogues in Table 2. Thiazole analogue 17a showed 83% ex vivo inhibition along with moderate microsomal stability against human and mouse liver microsomes (Table 2, 21 and 18 min, respectively). Thiophene analogues 18a (HSD-621)14 and 18b also displayed robust ex vivo inhibition in the adipose. As observed in the previous examples, the R-isomer (HSD-621) displayed a more desirable overall profile when compared to the S-isomer 18b. HSD-621 demonstrated more potent inhibition against human 11β-HSD1 (10 vs 145 nM), longer half-life when incubated with human microsomes (>30 vs 18 min), and more robust inhibition in the mouse epi fat ex vivo assays when orally dosed at 10 mg/kg (75 vs 50%).
The crystal structure of HSD-621 presents a “reversed” binding mode as predicted with the carboxamide on the thiophene side of the molecule directly interacting with the NADP through a hydrogen bond with the phosphate while maintaining a hydrogen bond with backbone A217 (O–N: 3.3 Å). The amide carbonyl oxygen of HSD-621 interacts with Y183 OH (Figure 3; O–O: 2.8 Å), and the amide NH forms a hydrogen bond with the phosphate oxygen (N–O: 3.3 Å). In this “reversed” binding mode, the lipophilic binding pocket A is satisfied by the trifluoromethyl group (CF3) at the C5 position of the thiophene ring. 2-Trifluoromethyl-4-fluorophenyl group now interacts with Y177 (edge-to-face π-stacking) and P178 as well as the C-terminal helix of the dimer partner chain.
Figure 3.
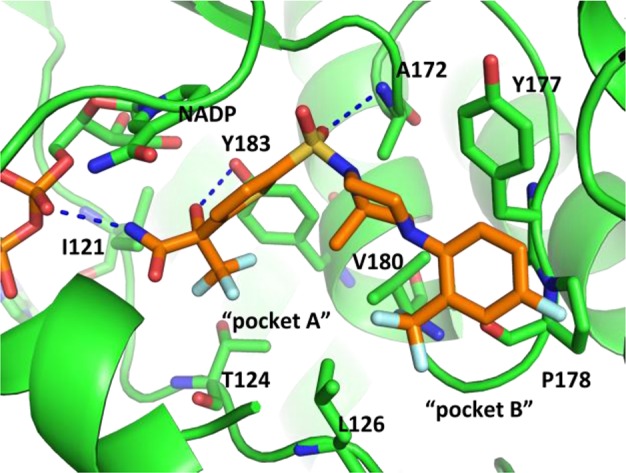
Crystal structure of HSD-621 bound to human 11β-HSD1. The inhibitor is shown with yellow carbons, and the NADP cofactor and residues containing atoms within 4 Å of the inhibitor are shown in green. The figure was generated with Pymol.
A number of analogues bearing amide isosteres were designed and synthesized as exemplified in compound 19. The cell-based activity and mouse epididymal fat ex vivo activity were comparable to those of HSD-621 (Table 2). Unfortunately, this compound had poor rat liver microsomal stability.
As shown in Tables 1 and 2, compounds 9a, 16a, and HSD-621 have good cell-based activity, microsomal stability, and epididymal fat ex vivo potency. Selectivity over 11β-HSD2 was also found to be excellent with IC50 values greater than 100 μM for all three compounds. These compounds were advanced into pharmacokinetic (PK) studies as summarized in Table 3.
Table 3. Selected PK Profile of Compounds 9a, 16a, and HSD-621.
| iva |
poa |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd | species | Cl (mL/min/kg) | Vss (L/kg) | AUC (h ng/mL) | t1/2 (h) | Cmax (ng/mL) | tmax (h) | AUC (h ng/mL) | t1/2 (h) | F (%) |
| 9a | mouseb | 9.0 | 5.8 | 3588 | 9.5 | 449 | 0.5 | 3403 | 6.0 | 19 |
| 16a | mouseb | 5.4 | 1.8 | 6131 | 4.2 | 1554 | 2.0 | 14005 | 2.8 | 46 |
| HSD-621 | mouseb | 9.0 | 4.2 | 3600 | 6.1 | 987 | 2.0 | 11700 | 4.8 | 67 |
| HSD-621 | ratc | 8.0 | 3.8 | 4310 | 8.6 | 656 | 1.8 | 6542 | 3.3 | 61 |
| HSD-621 | dogd | 1.0 | 1.8 | 32358 | 20.3 | 1444 | 2.8 | 43488 | 17.2 | 54 |
See the Experimental Procedures for dosing and analytical protocols.
C57B6 mice: iv dosed at 2 mg/kg in 50% DMSO/50% PEG200 and po dosed at 10 mg/kg in 2% Tween/0.5% methyl cellulose in water.
SD rat: iv dosed at 2 mg/kg in 50% DMSO/50% PEG200 and po dosed at 10 mg/kg in 2% Tween/0.5% methyl cellulose in water.
Male dog: iv dosed at 2 mg/kg in 50% DMSO/50% PEG200 and po dosed at 5 mg/kg in 2% Tween/0.5% methyl cellulose in water.
In a side-by-side comparison in the male C57B6 mouse PK studies, compound HSD-621 demonstrated a more balanced overall PK profile than compounds 9a and 16a. This compound has a low clearance and good in vivo half-life and oral bioavailability (Table 3, 67%). Therefore, HSD-621 was taken forward into further PK studies in male Sprague–Dawley (SD) rats and male beagle dogs—the species used for safety evaluations. As illustrated in Table 3, clearance of HSD-621 was low as compared to the hepatic blood flow rate in these animal species, resulting in a long t1/2. The oral bioavailability was greater than 50% in both species tested (Table 3, 61 and 54%, respectively).
Because of its desirable overall pharmaceutical profile, HSD-621 was selected for further safety screens. In addition to its selectivity over 11β-HSD2, excellent selectivity against peroxisome proliferator-activated receptors (PPAR) α, β, and γ was also observed (>100 μM). In the in vitro metabolism studies, there were no major metabolites detected, and half-lives were >30 min in rat, mouse, and human liver microsomes. No the human ether-à-go-go-related gene (hERG) ion channel activity was observed at 33 μM. HSD-621 was also clean in genotoxicity assay (AMES test) and clean in the nova screen against 134 targets at 5 μM. In 14 day safety assessment and metabolism studies, HSD-621 was well tolerated in rats dosed at 200, 600, and 2000 mg/kg and dogs dosed at 100, 300, and 1000 mg/kg.
To evaluate the in vivo efficacy of HSD-621, we administered this compound to male C57B6 mice (5.5–7 months old and on a 35.5% fat diet for 4 months) mixed in food at 0.3 mg drug/g food. This dose provides an AUC equivalent to that of an oral dosing at 2 mg/kg. HSD-621 treatment of C57B6 DIO mice for 31 days led to 19% reduction in fed glucose, 44% reduction in fed insulin (Figure 4), 15% reduction in fasting glucose, and 33% reduction in fasting insulin levels. No abnormalities were observed during the course or at the end of the study. There was no body weight loss, no reduction of food intake, and no elevation of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels. It was also noticed that no weight gain was observed throughout the study as compared to the control group or to the group treated with rosiglitazone at 0.25 mg drug/g food.
Figure 4.
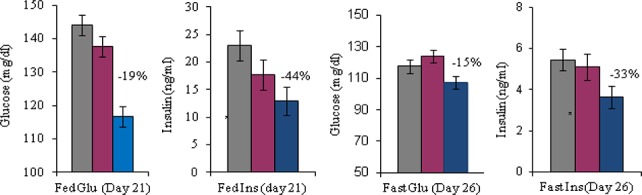
In vivo studies of HSD-621 (P < 0.05, see the Experimental Procedures for assay protocols). Note that diet 12492 was used as a control (gray color). Dose: 0.1 mg of HSD-621/g food (red color) and 0.3 mg of HSD-621/g food (blue color).
In summary, we developed a series of (R)-2-methylpiperazine sulfonamides as potent and selective 11β-HSD1 inhibitors. This led to the identification of HSD-621, a potent, selective, and orally efficacious 11β-HSD1 inhibitor with excellent pharmacokinetic and safety properties suitable for advancement into human clinical trials.
Acknowledgments
We thank Nelson Huang, Ning Pan, Peter Tate, Walter Massefski, and Li Di for coordinating and obtaining analytical data. We also thank Dr. Katherine Lee for proofreading this manuscript.
Supporting Information Available
Biological assays and experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Ozoles J. Lumenal Orientation and Post-translational Modifications of the Liver Microsomal 11β-Hydroxysteroid Dehydrogenase. J. Biol. Chem. 1995, 270, 2305–2312. [DOI] [PubMed] [Google Scholar]
- Odermatt A.; Arnold P.; Stauffer A.; Frey B. M.; Frey F. J. J. Biol. Chem. 1999, 274, 28762–28770. [DOI] [PubMed] [Google Scholar]
- Seckl J. R.; Walker B. R. 11β-Hydroxysteroid dehydrogenase type 1-a tissue-specific amplifier of glucocorticoid action. Endocrinology 2001, 142, 1371–1376. [DOI] [PubMed] [Google Scholar]
- Stulnig T. M.; Waldhausl W. 11β-Hydroxysteroid dehydrogenase type 1 in obesity and type 2 diabetes. Diabetologia 2004, 47, 1–11. [DOI] [PubMed] [Google Scholar]
- For a few selected reviews, seeWalker B. R.; Seckl J. R. 11β-Hydroxysteroid dehydrogenase type 1 as a novel therapeutic target in metabolic and neurodegenerative disease. Expert Opin. Ther. Targets 2003, 7, 771–783. [DOI] [PubMed] [Google Scholar]
- Tomlinson J. W.; Walker E. A.; Bujalska I. J.; Draper N.; Lavery G. G.; Cooper M. S.; Hewison M.; Stewart P. M. 11β-Hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [DOI] [PubMed] [Google Scholar]
- Hollis G.; Huber R. 11β-Hydroxysteroid dehydrogenase type 1 inhibition in type 2 diabetes mellitus. Diabetes, Obes. Metab. 2010, 13, 1–6. [DOI] [PubMed] [Google Scholar]
- Hosfield D. J.; Wu Y.; Skene R. J.; Hilgers M.; Jennings A.; Snell G. P.; Aertgeerts K. Conformational flexibility in crystal structures of human 11β-hydroxysteroid dehydrogenase type I provide insights into glucocorticoid interconversion and enzyme regulation. J. Biol. Chem. 2005, 280, 4639–4648. [DOI] [PubMed] [Google Scholar]
- Draper N.; Walker E. A.; Bujalska I. J.; Tomlinson J. W.; Chalder S. M.; Arlt W.; Lavery G. G.; Bedendo O.; Ray D. W.; Laing I.; Malunowicz E.; White P. C.; Hewison M.; Mason P. J.; Connell J. M.; Shackleton C. H.; Stewart P. M. Nat. Genet. 2003, 34, 434–439. [DOI] [PubMed] [Google Scholar]
- Albiston A. L.; Obeyesekere V., R.; Smith R. E.; Krozowski Z. S. Cloning and tissue distribution of human 11β-hydroxysteroid dehydrogenase type 2 enzyme. Mol. Cell Endocrinol. 1994, 105, R11–R17. [DOI] [PubMed] [Google Scholar]
- Masuzaki H.; Peterson J.; Shinyama H.; Morton N. M.; Mullins J. J.; Seckl J. R.; Flier J. S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [DOI] [PubMed] [Google Scholar]
- Masuzaki H.; Yamamoto H.; Kenyon C. J.; Elmquist J. K.; Morton N. M.; Paterson M. M.; Shinyama H.; Sharp M. G.; Fleming S.; Mullins J. J.; Seckl J. R.; Flier J. S. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J. Clin. Invest. 2003, 112, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotelevtsev Y.; Holmes M. C.; Burchell A.; Houston P. M.; Schmoll D.; Jamieson P.; Best R.; Vrown R.; Edwards C. R.; Seckl J. R.; Mullins J. J. 11 Beta–hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 14924–14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton N. M.; Paterson M.; Masuzaki H.; Holmes M.; Staels B.; Fievet C.; Walker B.; Flier J. S.; Mullins J. J.; Seckl J. R. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11β-hydroxysteroid dehydrogenase type-1 deficient mice. Diabetes 2004, 53, 931–938. [DOI] [PubMed] [Google Scholar]
- Stimson R. H.; Andrew R.; McAvoy N. C.; Tripathi D.; Hayes P. C.; Walker B. R. Increased whole-body and sustained liver cortisol regeneration by 11β-hydroxysteroid dehydrogenase type 1 in obese men with type 2 diabetes provides a target for enzyme inhibition. Diabetes 2011, 60, 720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selected recent reports on 11β-HSD1 inhibitors:; Xiang J.; Wan Z.-K.; Li H.-Q.; Ipek M.; Binnun E.; Nunez J.; Chen L.; McKew J. C.; Mansour T. S.; Xu X.; Suri V.; Tam M.; Xing Y.; Li X.; Hahm S.; Tobin J.; Saiah E. Piperazine sulfonamides as potent, selective, and orally available 11β-hydroxysteroid dehydrogenase type 1 inhibitors with efficacy in the rat cortisone-induced hyperinsulinemia model. J. Med. Chem. 2008, 51, 4068–4071. [DOI] [PubMed] [Google Scholar]
- Wan Z.-K.; Chenail E.; Xiang J.; Li H.-Q.; Ipek M.; Bard J.; Svenson K.; Mansour T. S.; Xu X.; Tian X.; Suri V.; Hahm S.; Xing Y.; Johnson C. E.; Li X.; Qadri A.; Panza D.; Perreault M.; Tobin J.; Saiah E. Efficacious 11β-hydroxysteroid dehydrogenase type 1 inhibitors in the diet-induced obesity mouse model. J. Med. Chem. 2009, 52, 5449–5461. [DOI] [PubMed] [Google Scholar]
- Su X.; Pradaux-Caggiano F.; Thomas M. P.; Szeto M.W. Y.; Halem H. A.; Culler M. D.; Vicker N.; Potter B. V. L Discovery of adamantyl ethanone derivatives as potent 11β-hydroxysteroid dehydrogenase Type 1 (11β-HSD1) Inhibitors. ChemMedChem 2010, 5, 1026–1044. [DOI] [PubMed] [Google Scholar]
- Veniant M. M.; Hale C.; Hungate R. W.; Gahm K.; Emery M. G.; Jona J.; Joseph S.; Adams J.; Hague A.; Moniz G.; Zhang J.; Bartberger M. D.; Li V.; Syed R.; Jordan S.; Komorowski R.; Chen M. M.; Cupples R.; Kim K. W.; St. Jean D. J. Jr.; Johansson L.; Henriksson M. A.; Williams M.; Vallgarda J.; Fotsch C.; Wang M. Discovery of a potent, orally active 11β-hydroxysteroid dehydrogenase type 1 inhibitor for clinical study: Identification of (S)-2-((1S,2S,4R)-bicyclo[2.2.1]heptan-2-ylamino)-5-isopropyl-5-methylthiazol-4(5H)-one (AMG 221). J. Med. Chem. 2010, 53, 4481–4487. [DOI] [PubMed] [Google Scholar]
- Torres-Piedra M.; Ortiz-Andrade R.; Villalobos-Molina R.; Singh N.; Medina-Franco J. L.; Webster S. P.; Binnie M.; Navarrete-Vazquez G.; Estrada-Soto S. A comparative study of flavonoid analogues on streptozotocin-nicotinamide induced diabetic rats: Quercetin as a potential antidiabetic agent acting via 11β-Hydroxysteroid dehydrogenase type 1 inhibition. Eur. J. Med. Chem. 2010, 45, 2606–2612. [DOI] [PubMed] [Google Scholar]
- Rollinger J. M; Kratschmar D. V; Schuster D.; Pfisterer P. H; Gumy C.; Aubry E. M; Brandstotter S.; Stuppner H.; Wolber G.; Odermatt A. 11beta-Hydroxysteroid dehydrogenase 1 inhibiting constituents from Eriobotrya japonica revealed by bioactivity-guided isolation and computational approaches. Bioorg. Med. Chem. 2010, 18, 1507–15. [DOI] [PubMed] [Google Scholar]
- Venier O.; Pascal C.; Braun A.; Namane C.; Mougenot P.; Crespin O.; Pacquet F.; Mougenot C.; Monseau C.; Onofri B.; Dadji-Faihun R.; Leger C.; Ben-Hassine M.; Van-Pham T.; Ragot J.-L.; Philippo C.; Guessregen S.; Engel C.; Farjot G.; Noah L.; Maniani K.; Nicolai E. Pyrrolidine-pyrazole ureas as potent and selective inhibitors of 11β-hydroxysteroid-dehydrogenase type 1. Bioorg. Med. Chem. Lett. 2011, 21, 2244–2251. [DOI] [PubMed] [Google Scholar]
- Sun W.; Maletic M.; Mundt S. S.; Shah K.; Zokian H.; Lyons K.; Waddell S. T.; Balkovec J. Substituted phenyl triazoles as selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1. Bioorg. Med. Chem. Lett. 2011, 21, 2141–2145. [DOI] [PubMed] [Google Scholar]
- Li A.; Yuan C. C.; Chow D.; Chen M.; Emery M. G.; Hale C.; Zhang X.; Subramanian R.; St. Jean D. Jr.; Komorowski R.; Veniant M.; Wang M.; Fostsch C. Synthesis and Evaluation of the Metabolites of AMG 221, a Clinical Candidate for the Treatment of Type 2 Diabetes. ACS Med. Chem. Lett. 2011, 2, 824–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X.-Y.; Chen S. Y.; Nayeem A.; Golla R.; Seethala R.; Wang M.; Harper T.; Sleczka B. G.; Li Y.-X.; He B.; Kirby M.; Gordon D. A.; Robl J. A. Design, synthesis, and SAR studies of novel polycyclic acids as potent and selective inhibitors of human 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD-1). Bioorg. Med. Chem. Lett. 2011, 21, 6669–6704. [DOI] [PubMed] [Google Scholar]
- Scott J. S.; Bowker S. S.; deSchoolmeester J.; Gerhardt S.; Hargreaves D.; Kilgour E.; Lloyd A.; Mayers R. M.; McCoull W.; Newcombe N. J.; Ogg D.; Packer M. J.; Rees A.; Revill J.; Schofield P.; Selmi N.; Swales J. G.; Whittamore P. R. O. J. Med. Chem. 2012, 55, 5951–5964. [DOI] [PubMed] [Google Scholar]
- Barf T.; Vallgarda J.; Emond R.; Haggstrom C.; Kurz G.; Nygren A.; Larwood V.; Mosialou E.; Axelsson K.; Olsson R.; Engblom L.; Edling N.; Ronquist-Nii Y.; Ohman B.; Alberts P.; Abrahmsen L. J. Med. Chem. 2002, 45, 3813–3815. [DOI] [PubMed] [Google Scholar]
- Alberts P.; Engblom L.; Edling N.; Forsgren M.; Klingstrom G.; Larsson C.; Ronquist-Nii Y.; Ohman B.; Abrahmsen L. Diabetologia 2002, 45, 1528. [DOI] [PubMed] [Google Scholar]
- Alberts P.; Nilsson C.; Selen G.; Engblom L. O.; Edling N. H.; Norling S.; Klingstrom G.; Larsson C.; Forsgren M.; Ashkzari M.; Nilsson C. E.; Fiedler M.; Bergqvist E.; Ohman B.; Bjorkstrand E.; Abrahmsen L. B. Endocrinology 2003, 144, 4755. [DOI] [PubMed] [Google Scholar]
- Hermanowski-Vosatka A.; Balkovec J. M.; Cheng K.; Chen H. Y.; Hernandez G. C.; Koo G. C.; LeGrand C. B.; Li Z.; Metzger M. M.; Mundt S. S.; Noonan H.; Nunes C. N.; Olson S. H.; Pikounis B.; Ren N.; Robertson N.; Schaeffer M. M.; Shah K.; Springer M. S.; Strack A. M.; Strowski M.; Wu K.; Wu T.; Xiao J.; Zhang B. B.; Wright S. D.; Thieringer R. J. Exp. Med. 2005, 202, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat B. G.; Hosea N.; Fanjul A.; Herrera J.; Chapman J.; Thalacker F.; Stewart P. M.; Rejto P. A. Demonstration of proof of mechanism and pharmacokinetics and pharmacodynamic relationship with 4′-cyano-biphenyl-4-sulfonic acid (6-amino-pyridin-2-yl)-amide (PF-915275), an inhibitor of 11β-hydroxysteroid dehydrogenase type 1, in cynomolgus monkeys. J. Pharmacol. Exp. Ther. 2008, 324, 299–305. [DOI] [PubMed] [Google Scholar]
- Rosenstock J.; Banarer S.; Fonseca V. A.; Inzucchi S. E.; Sun W.; Yao W.; Hollis G.; Flores R.; Levy R.; Williams W. V.; Seckl J. R.; Huber R. The 11β-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 2010, 33, 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson J. M.; Morton N. M.; Fievet C.; Kenyon C. J.; Holmes M. C.; Staels B.; Seckl J. R.; Mullins J. J. Metabolic syndrome without obesity: Hepatic overexpression of 11β-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 7088–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Z.-K.; Chenail E.; Li H.-Q.; Kendall C.; Wang Y.; Gingras S.; Xiang J.; Massefski W.; Mansour T. S.; Saiah E. Synthesis of potent and efficacious 11β-hydroxysteriod dehydrogenase type I inhibitor HSD-016. J. Org. Chem. 2011, 76, 7048–7055. [DOI] [PubMed] [Google Scholar]
- Aahni-Arya B.; Flynn M. J.; Bergeron L.; Salyan M. E. K.; Pedicord D. L.; Golla R.; Ma Z.; Wang H.; Seethala R.; Wu S. C.; Li J. J.; Nayeem A.; Gates C.; Hamann L. G.; Gordon D. A.; Blat Y. Cofactor-specific modulation of 11β-hydroxysteroid dehydrogenase 1 inhibitor potency. Biochim. Biophys. Acta 2007, 1774, 1184–1191. [DOI] [PubMed] [Google Scholar]
- The absolute stereochemistry of 18a (HSD-621) is assigned by single-crystal X-ray (see the coordinates table in the Supporting Information).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.