

Abstract
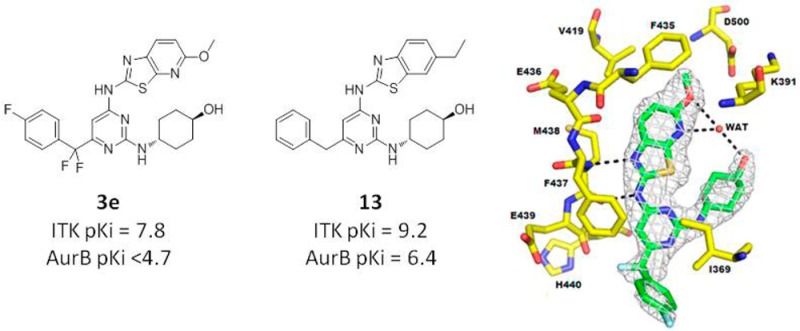
Inhibition of Itk potentially constitutes a novel, nonsteroidal treatment for asthma and other T-cell mediated diseases. In-house kinase cross-screening resulted in the identification of an aminopyrazole-based series of Itk inhibitors. Initial work on this series highlighted selectivity issues with several other kinases, particularly AurA and AurB. A template-hopping strategy was used to identify a series of aminobenzothiazole Itk inhibitors, which utilized an inherently more selective hinge binding motif. Crystallography and modeling were used to rationalize the observed selectivity. Initial exploration of the SAR around this series identified potent Itk inhibitors in both enzyme and cellular assays.
Keywords: Interleukin-2 inducible tyrosine kinase, Itk, kinase inhibitors, aminobenzothiazole, template hopping, kinase selectivity
Interleukin-2 inducible tyrosine kinase (Itk) is a nonreceptor protein tyrosine kinase that is expressed in T cells, mast cells, and NK cells. Itk plays an important role in signaling, downstream of the T cell receptor in response to antigen presentation by MHC proteins, and its inhibition leads to reduced levels of key inflammatory cytokines.1 In vivo experiments with Itk knockout mice suggest a role for Itk inhibitors in the treatment of asthma.2
A number of Itk inhibitor series have been disclosed in the literature with a focus on achieving broad kinase selectivity as well as good levels of cellular activity; both of which have been relatively challenging for this tyrosine kinase.3−6 Despite these publications, there have been no reports of an Itk inhibitor entering clinical trials and hypotheses regarding its clinical potential remain untested.7
In-house cross screening resulted in the identification of a series of aminopyrazoles as inhibitors of Itk. This series was of particular interest to us as, in contrast to previous series investigated, compounds in this series displayed a promising level of ligand efficiency (LE = 0.36, compound 1).8 An initial X-ray crystal structure of compound 1 (Figure 1) in Itk confirmed the aminopyrazole group was binding to the hinge region of Itk and utilizing a three-point hinge binding motif. Initial optimization work focused on the pyrimidine 2-position and the pendant group of the pyrazole. However, these modifications did not produce compounds with the desired 100-fold selectivity margin over key kinases, namely, LCK, AurA, and AurB. Compound 2 (Figure 1) represents the best combination of potency and selectivity achieved with this series. Substantial SAR knowledge had been built up around the other parts of the template at this stage, and it was hypothesized that replacing the aminopyrazole motif with an inherently more selective hinge-binder could be an efficient method of accessing novel and selective Itk inhibitors.
Figure 1.

Structure and activity of aminopyrazole-based Itk inhibitors. Activity data presented as pKi.13
A robust Itk crystallography system was not available at this time, and therefore a fragment based approach9−11 using crystallography to identify new hinge-binding groups was not feasible. Instead, two different methods for selecting alternative hinge-binders were utilized. The first of these used an in-house set of compounds specifically chosen for their potential to bind to the hinge region of a kinase.12 This set was screened against Itk and any hits selected for use in this work.
Additionally, a set of low molecular weight hits from an historical high-throughput screen, together with ongoing cross-screening hits that had not yet been followed-up, were re-examined. Hits of interest were rescreened at higher compound concentration if necessary. This set of hits was analyzed to identify hinge-binders of interest. Together, these methods enabled us to select a set of aminoheterocycles to act as replacement hinge-binders.
A set of compounds was designed to rapidly evaluate the potential of these new hinge-binders. The central pyrimidine ring of the original template was retained although the 6-benzyl group was replaced with the fluorinated derivative (Figure 2) which was tolerated with minimal loss of potency and greatly contributed to synthetic ease. The aminoheterocycles chosen to act as replacement hinge-binders were introduced in the 4-position and each hinge-binder was synthesized as both the trans-aminocyclohexanol and l-prolineamide analogues (Figure 2). These moieties had produced the best overall results in the aminopyrazole series.
Figure 2.

General structure of hinge-binding replacement set.
This set was synthesized by the general method shown in Scheme 1. Methyl 2,6-dichloropyrimidine-4-carboxylate 18 was treated with 4-fluorophenylmagnesium bromide, and the resulting ketone 7 reacted with DAST to yield the general pyrimidine intermediate 19.14 Reaction with the chosen aminoheterocycles was typically carried out in the presence of sodium hydride, and each intermediate 21 was reacted separately with both trans-aminocyclohexanol and l-prolineamide using microwave heating in the presence of DIPEA.15
Scheme 1.

The potency and selectivity criteria set at the start of this work were a pKi of at least 7.5 in the Itk enzyme assay combined with a minimum 100-fold selectivity over AurA and AurB (due to their fundamental role in cell cycle regulation16). Potential for further optimization was also a desirable quality. Some key results from this set of compounds are shown in Table 1. A variety of different heterocycles were targeted as replacement hinge-binders. Most of these had some activity at Itk although many were at least as active at Aurora (e.g., 3a, Table 1). Interestingly, it was observed that the SAR from the aminopyrazole series was not always transferable to this set of compounds. Compounds 3d and 4d (Table 1) are direct analogues that differ only in the group at the 2-position. The trans-aminocyclohexanol analogue meets the potency criteria, while the prolineamide analogue is inactive in the Itk assay. More encouraging results were obtained with benzothiazole-based inhibitors. Both the benzothiazole 3f and aza-benzothiazole 3e analogues fulfilled the potency and selectivity criteria that were set at the start of the work. The l-prolineamide analogue 4f was 100-fold less active.
Table 1. Replacement Hinge-Binder Compound Set, Activity, and Selectivity Data.
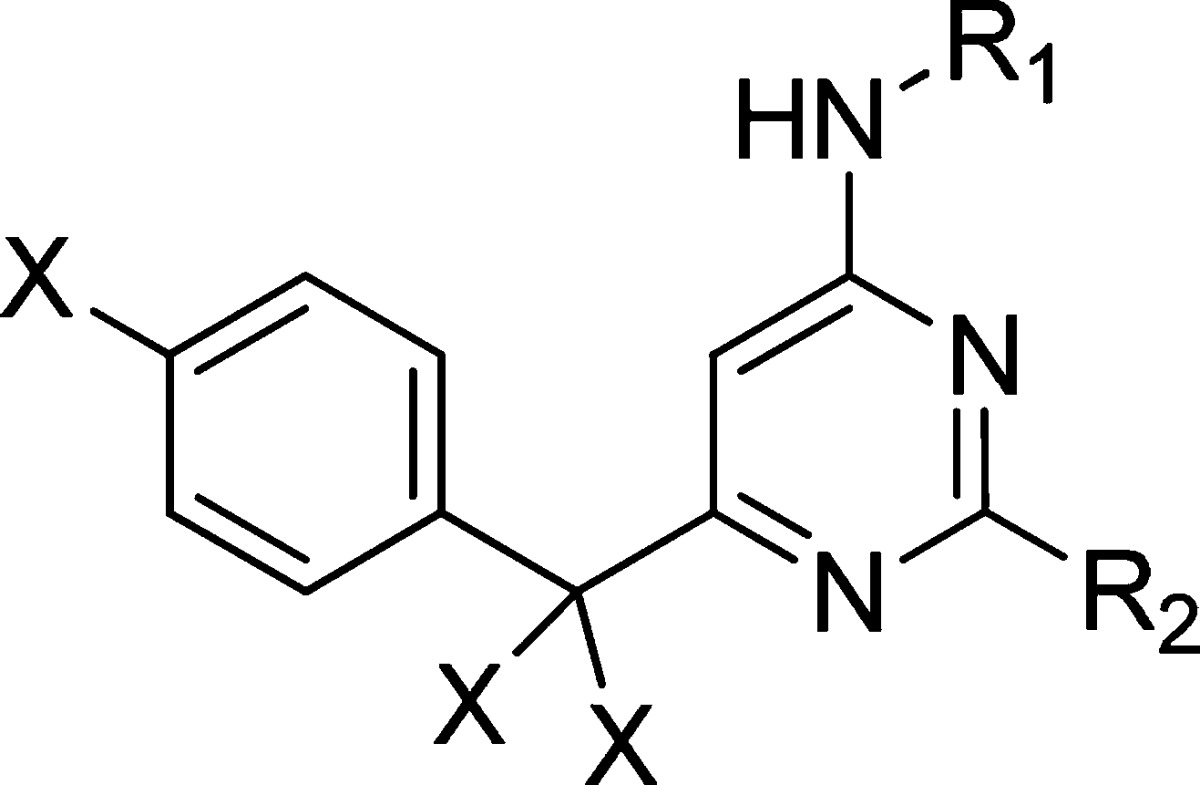
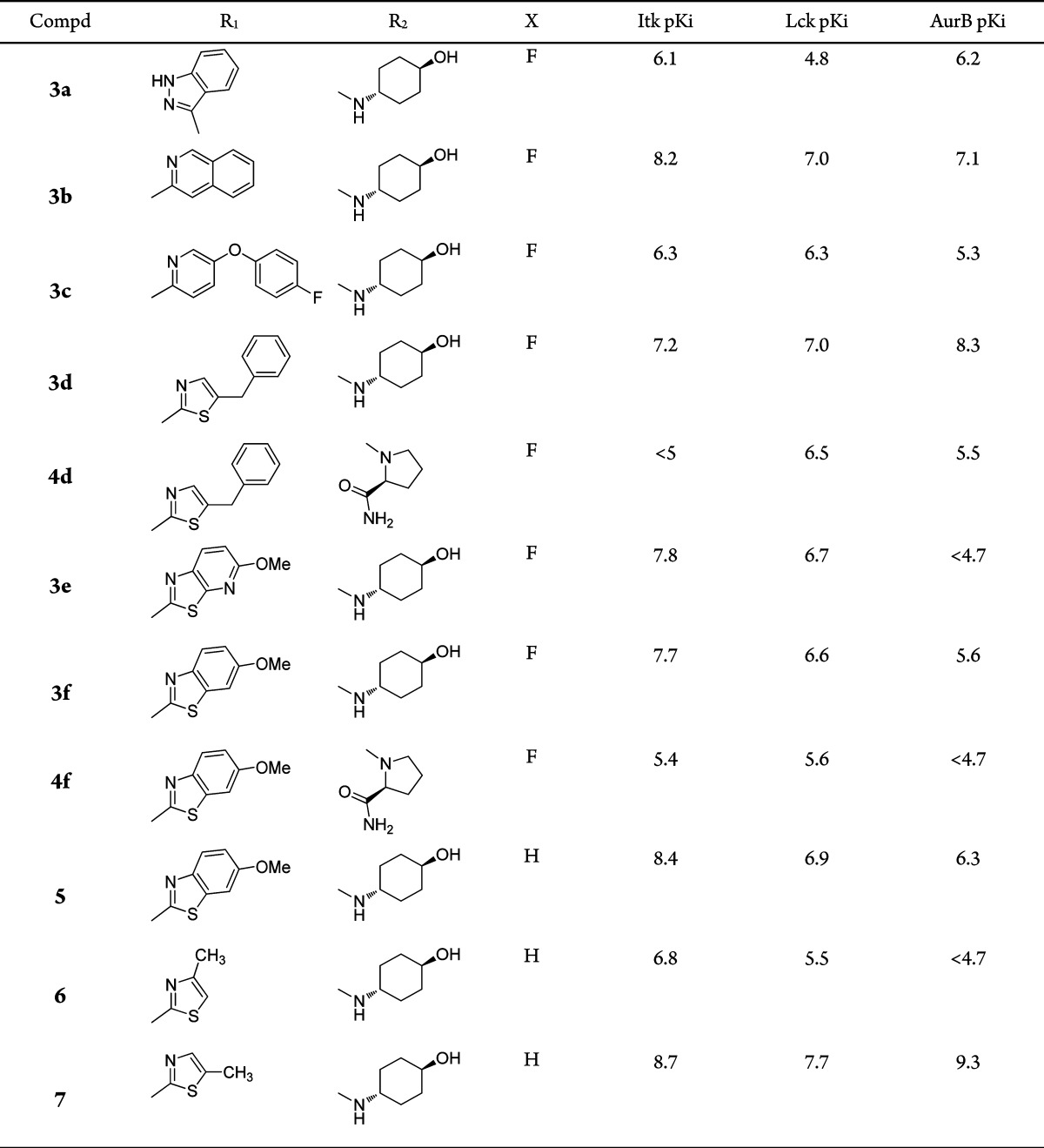
From our work on the aminopyrazole series, it seemed likely that the des-fluoro analogue of compound 3f would demonstrate an increase in Itk potency. This was indeed the case with a 5-fold increase in potency observed (compound 5, Table 1), and gratifyingly, it also retained the selectivity window over AurA and AurB.17 Although selectivity over Lck was not ideal at this stage, it was perceived to be a solvable issue. The Aurora selectivity was the key attribute that had not been demonstrated with previous series. These benzothiazole-based inhibitors also demonstrated encouraging cellular data: compound 5 achieved a pIC50 of 7.6 in a cell assay measuring inhibition of IFNγ production from PBMCs.
The next step was to rationalize the Aurora selectivity observed with the aim of increasing Itk potency while maintaining Aurora selectivity. Without the benefit of a crystal structure of a compound from this series at the time, we used computational models derived from available Itk and Aurora crystal structures to guide us. Compound 3f was docked into Itk (GLIDE SP, Schrodinger Inc.19), showing clearly that the C4–C5 region of the benzothiazole/thiazolopyridine ring fitted well against Val419, adjacent to the gatekeeper residue, Phe435, while maintaining a strong hinge-binding motif. It was observed that the equivalent residue in Aurora (Leu194) protrudes into the space occupied by C4–C5 as docked into Itk. Examination of previous models of nonselective inhibitors showed that hitherto this critical space in the receptor had not been occupied. It was hypothesized that the steric clash between the benzothiazole/thiazolopyridine C4–C5 and Aurora’s Leu194 side chain could explain the observed selectivity of this series. The model was used to design a small set of aminothiazole analogues to test our hypothesis. Compounds that presented a methyl group at position 4 of the thiazole (e.g., compound 6), equivalent to the benzothiazole/thiazolopyridine C4, exhibited the same significant selectivity for Itk over Aurora. Absence of a 4-position substituent abolished all selectivity (e.g., compound 7).
It was now apparent that an inherently more selective hinge-binder had been identified. The next step was to explore substitution around the benzothiazole ring and scope out the initial SAR. At this time, because of the increasing Itk activity of this series, it was necessary to reconfigure the assay to run with a higher concentration of the substrate ATP.20,21 This increased competition with inhibitors of the ATP binding site and effectively shifted the pKi range of the assay upward making it possible to differentiate between our most potent compounds. Substitution at the 5-position resulted in a drop-off in potency at Itk (Table 2, compound 8), adding further weight to our theory that the benzothiazole core is a close fit against the kinase surface in Itk. Limited work was carried out at the 7-position at this time, although it was observed that replacement of hydrogen with bromine did not result in any significant change in Itk potency. The 6-position was explored in more detail, and it was observed that a range of substituents were tolerated at this position (compounds 10–14) the highlight being the 6-ethyl analogue 13 (LE = 0.38), which was inactive against AurA and highly selective over AurB. Switching to the thiazolopyridine core (compounds 15–17) generally resulted in compounds that maintained their Itk potency and selectivity over Lck, AurA, and AurB.
Table 2. Representative SAR and Benzothiazole Ring System.
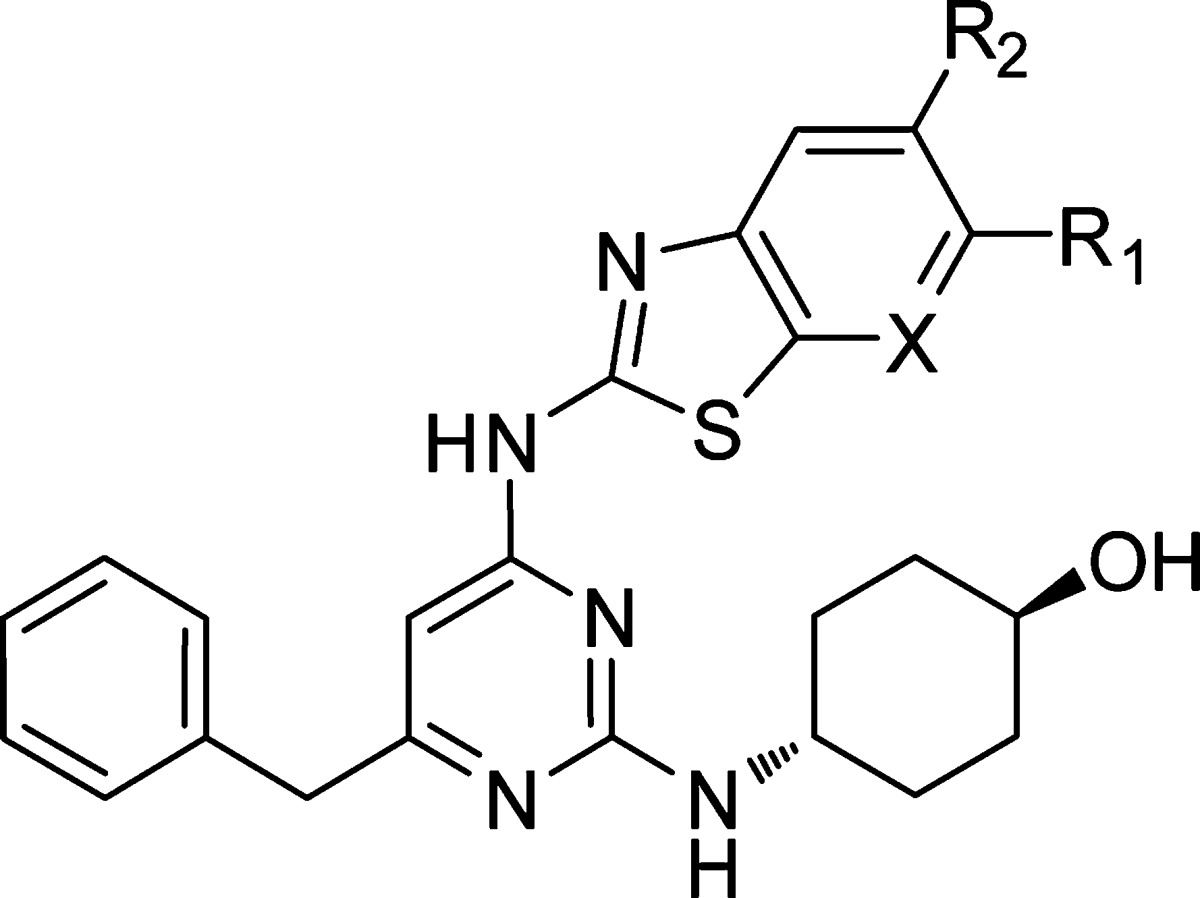
| compd | R1 | R2 | X | Itk pKi | Lck pKi | AurB pKi |
|---|---|---|---|---|---|---|
| 8 | Me | Me | C | <5a | <4.6 | 4.7 |
| 9 | H | H | C | 9.0 | 6.5 | 6.4 |
| 10 | Me | H | C | 9.4 | 6.6 | 6.5 |
| 11 | Cl | H | C | 9.5 | 7.1 | 6.6 |
| 12 | CF3 | H | C | 7.9a | 7.3 | 6.6 |
| 13 | Et | H | C | 9.2 | 7.0 | 6.4 |
| 14 | CN | H | C | 9.6 | 7.2 | 7.1 |
| 15 | OMe | H | N | 9.7 | 7.1 | 6.5 |
| 16 | H | H | N | 9.6 | 7.0 | 7.0 |
| 17 | Et | H | N | 8.8 | 7.0 | 7.2 |
pKi measured at low ATP concentration.21
Compound 13 had demonstrated the best selectivity against a limited in-house panel of kinases and hence was chosen as the series exemplar for profiling against a wider panel of kinases (Table 3). After further in-house screening and profiling at Upstate,22 compound 13 was found to inhibit only 3 other kinases at a pIC50 greater than 7. Importantly, in addition to the desired selectivity over Aurora,23 this compound was also sufficiently selective over the kinase Btk, which is closely related to Itk and part of the same TEC family of kinases. The main selectivity issue with this compound (and this series) was the activity observed against IRAK4, Lck, and related kinase Src. Compound 13 also achieved a pIC50 of 7.3 in the cell assay, measuring inhibition of IFNγ production from PBMCs.
Table 3. Kinase Selectivity Profile of Compound 13.
| kinase | enzyme pIC50 | kinase | enzyme pIC50 |
|---|---|---|---|
| IRAK4 | 8.4 | CAMKK2 | 6.7 |
| Src | 7.4 | Tec | 6.6 |
| TXK | 7.1 | Btk | 6.5 |
| Lck | 6.9 | LRRK2 | 6.4 |
| Bmx | 6.8 | AurB | 6.4 |
| IR | 6.8 | EGFR | 6.2 |
At this time, the 2.0 Å resolution cocrystal structure of Itk in complex with compound 3e, one of our initial thiazolopyridine hits, was obtained (Figure 3a).24 Compound 3e binds in the ATP-binding domain of Itk; the aminothiazolopyridine core makes two H-bonding interactions with the backbone carbonyl and NH of Met438 in the hinge region and is flanked by the side chains of Ala389 and Leu489. Compound 3e appears to adopt a conformation stabilized by a water-bridged hydrogen-bonding network between the methoxy, pyridinyl, and cyclohexanol hydroxyl groups. The 6-methoxy group is located in a predominately hydrophobic subsite formed by gatekeeper Phe435, Val419, Ser499, and Asp500 of the DFG motif.
Figure 3.
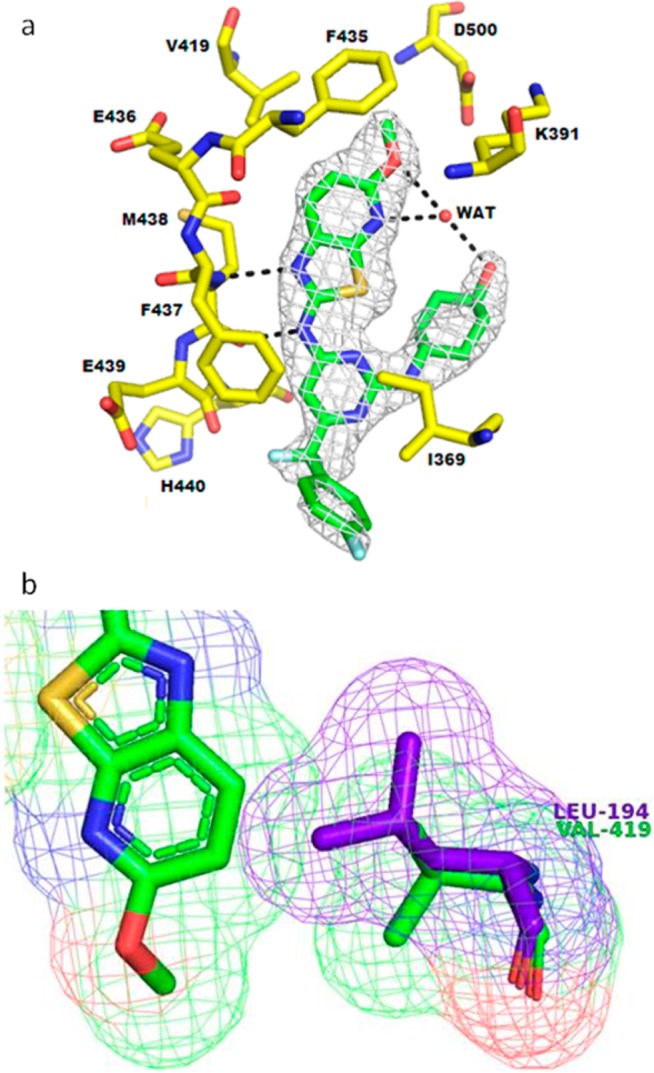
(a) Cocrystal structure of compound 3e bound to Itk kinase domain.24 Omit map electron density (at 3σ) is shown. (b) Connolly surfaces of compound 3e (green) and neighboring residue (Val419) of cocrystal structure overlaid with the equivalent AurA residue (Leu194, colored purple) defined from selected AurA crystal structures.25,26 This overlay highlights a possible reason for the improved selectivity of the Itk aminobenzothiazole inhibitor series with respect to Aurora.
The coplanar pyrimidinyl moiety resides within the space formed by Gly441 and the side chains of Ile369 and Phe437, directing the aminocyclohexanol moiety to a region where it stacks against the glycine-rich loop and Cys442. Additionally, there are long-range water mediated hydrogen bonds to C-terminal domain Asp445 side chain and Arg486 main chain carbonyl and the (moderately disordered) side chains of catalytic residues Lys391 and Asp500.
Modeling predicted this binding mode of compound 3e, and an overlay of Aurora A (Figure 3b) corroborated our view of the beneficial role of Val419 in Itk versus the larger Leu194 residue found at that position in AurA. It suggested that Leu194 would clash with the inhibitor, thus impeding binding and leading to the observed selectivity.
The crystal structure of 3e in Itk also highlighted the importance of Ser499, which is involved in a series of water mediated hydrogen bonds to the trans-aminocyclohexanol moiety. Analysis of 491 protein kinases revealed that serine is relatively rare at this position (less than 18% of kinases analyzed). Targeting this residue in order to further increase selectivity and potency was the next strategy that we explored, and the results of this work will form the basis of forthcoming publications.
In summary, we have identified a novel and selective series of Itk inhibitors using a template-hopping strategy. Excellent selectivity against key selectivity kinases AurA and AurB was achieved. Modeling and crystallography were instrumental in understanding the selectivity achieved against AurA and AurB and formulating strategies to further improve potency against Itk while retaining selectivity.
Glossary
Abbreviations
- ATP
adenosine triphosphate
- AurA
Aurora A
- AurB
Aurora B
- Btk
Bruton’s tyrosine kinase
- DAST
diethylaminosulfur trifluoride
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- IKKβ
inhibitor of nuclear factor kappa-β kinase subunit beta
- IPA
isopropyl alcohol
- IRAK4
Interleukin-1 receptor associated kinase 4
- Itk
interleukin-2 inducible tyrosine kinase
- LCK
lymphocyte-specific protein tyrosine kinase
- MHC
major histocompatibility complex
- PBMC
peripheral blood mononuclear cell
- SAR
structure–activity relationships
- TEC
tyrosine kinase expressed in hepatocellular carcinoma
- THF
tetrahydrofuran
Supporting Information Available
Experimental data for the synthesis and characterization of new compounds, assay protocols, and X-ray crystallography data. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The PDB accession code for the X-ray cocrystal structure of Itk + 3e is 4L7S.
Author Present Address
† (M.A.) MRC Technology, 1–3 Burtonhole Lane, Mill Hill, London, NW7 1AD, U.K.
Author Present Address
‡ (C.S.) University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, U.K.
Author Present Address
§ (C.W.) School of Life Sciences, Sir James Black Centre, University of Dundee, Dow Street, Dundee, DD1 5EH, U.K.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Grasis J. A.; Tsoukas C. D. Itk: The rheostat of the T cell response. J. Signal Transduction 2011, 267868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- August A.; Ragin M. J. Regulation of T-cell responses and diseases by Tec kinase Itk. Int. Rev. Immumol. 2012, 31, 155–165. [DOI] [PubMed] [Google Scholar]
- Das J.; Furch J. A.; Liu C.; Moquin R. V.; Lin J.; Spergel S. H.; McIntyre K. W.; Shuster D. J.; O’Day K. D.; Penhallow B.; Hung C. Y.; Doweyko A. M.; Kamath A.; Zhang H.; Marathe P.; Kanner S. B.; Lin T. A.; Dodd J. H.; Barrish J. C.; Wityak J. Discovery and SAR of 2-amino-5-(thioaryl)thiazoles as potent and selective Itk inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 3706–3712. [DOI] [PubMed] [Google Scholar]
- Riether D.; Zindell R.; Kowalski J. A.; Cook B. N.; Bentzien J.; Lombaert S. D.; Thomson D.; Kugler S. Z. Jr.; Skow D.; Martin L. S.; Raymond E. L.; Khine H. H.; O’Shea K.; Woska J. R. Jr.; Jeanfavre D.; Sellati R.; Ralph K. L.; Ahlberg J.; Labissiere G.; Kashem M. A.; Pullen S. S.; Takahashi H. 5-Aminomethylbenzimidazoles as potent ITK antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 1588–1591. [DOI] [PubMed] [Google Scholar]
- Charrier J. D.; Miller A.; Kay D. P.; Brenchley G.; Twin H. C.; Collier P. N.; Ramaya S.; Keily S. B.; Durrant S. J.; Knegtel R. M.; Tanner A. J.; Brown K.; Curnock A. P.; Jimenez J. M. Discovery and structure–activity relationship of 3-aminopyrid-2-ones as potent and selective interleukin-2 inducible T-cell kinase (Itk) inhibitors. J. Med. Chem. 2011, 54, 2341–2350. [DOI] [PubMed] [Google Scholar]
- Zapf C. W.; Gerstenberger B. S.; Xing L.; Limburg D. C.; Anderson D. R.; Caspers N.; Han S.; Aulabaugh A.; Kurumbail R.; Shakya S.; Li X.; Spaulding V.; Czerwinski R. M.; Seth N.; Medley Q. G. Covalent inhibitors of interleukin-2 inducible T-cell kinase (Itk) with nanomolar potency in a whole-blood assay. J. Med. Chem. 2012, 55, 10047–10063. [DOI] [PubMed] [Google Scholar]
- Lo H. Y. Itk inhibitors: a patent review. Expert Opin. Ther. Patents 2010, 20, 459–469. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; McDowell R. S.; O’Brien T. Fragment-based drug discovery. J. Med. Chem. 2004, 47, 3463–3482. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Chessari G.; Tisi D.; Woodhead A. J. Recent developments in fragment-based drug discovery. J. Med. Chem. 2008, 51, 3661–3680. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51, 4990–5003. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Brown M. J.; Christopher J. A.; Chung C.; Mellor G. W. Selectivity of kinase inhibitor fragments. J. Med. Chem. 2011, 54, 5131–5143. [DOI] [PubMed] [Google Scholar]
- Hancock A. A.; Bush E. N.; Stanisic D.; Kyncl J. J.; Lin C. T. Data normalization before statistical analysis: keeping the horse before the cart. Trends Pharmacol. Sci. 1998, 9, 29–32. [DOI] [PubMed] [Google Scholar]
- Fauq A. H.; Singh R. P.; Meshri D. T.. Encyclopedia of Reagents for Organic Synthesis; Wiley & Sons: New York, 2006. [Google Scholar]
- Alder C. M.; Baldwin I. R.; Barton N. P.; Campbell A. J.; Champigny A. C.; Harling J. D.; Maxwell A. C.; Simpson J. K.; Smith I. E. D.; Tame C. J.. Preparation of Pyrimidine Derivatives As Itk Kinase Inhibitors. WO 2010/106016 A1.
- Kelly K. R.; Ecsedy J.; Mahalingam D.; Nawrocki S. T.; Padmanabhan S.; Giles F. J.; Carew J. S. Targeting Aurora kinases in cancer treatment. Curr. Drug Targets 2011, 12, 2067–2078. [DOI] [PubMed] [Google Scholar]
- AurA and AurB share 71% identity within the catalytic domain18 and the key Leu residue is common to both. AurA and AurB activity tracked closely for this chemotype; AurB activity data was used to guide the medicinal chemistry strategy and hence is quoted here.
- Carmena M.; Earnshaw W. C. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell. Biol. 2003, 4, 842–854. [DOI] [PubMed] [Google Scholar]
- Glide, version 4.5; Schrödinger, LLC: New York, 2007.
- Cheng Y.; Prusoff W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Table 2 includes data from both standard (low ATP) and desensitized (high ATP) Itk assay formats as described in the Supporting Information. All other data described was generated using the standard assay format.
- Upstate kinase panel now part of Millipore; http://www.millipore.com/life_sciences/flx4/lead_discovery.
- Compound 13: Aur A pIC50 < 5.5.
- See Supporting Information for crystallographic details.
- Nowakowski J.; Cronin C. N.; McRee D. E.; Knuth M. W.; Nelson C. G.; Pavletich N. P.; Rogers J.; Sang B.-C.; Scheibe D. N.; Swanson R. V.; Thompson D. A. Structures of the cancer-related Aurora-A, FAK, and EphA2 protein kinases from nanovolume crystallography. Structure 2002, 10, 1659–1667. [DOI] [PubMed] [Google Scholar]
- Coumar M. S.; Leou J.-S.; Shukla P.; Wu J.-S.; Dixit A. K.; Lin W.-H.; Chang C.-Y.; Lien T.-W.; Tan U.-K.; Chen C.-H.; Hsu J. T-A.; Chao Y.-S.; Wu S.-Y.; Hsieh H.-P. Structure-based drug design of novel aurora kinase A inhibitors: Structural basis for potency and specificity. J. Med. Chem. 2009, 52, 1050–1062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.