

Abstract
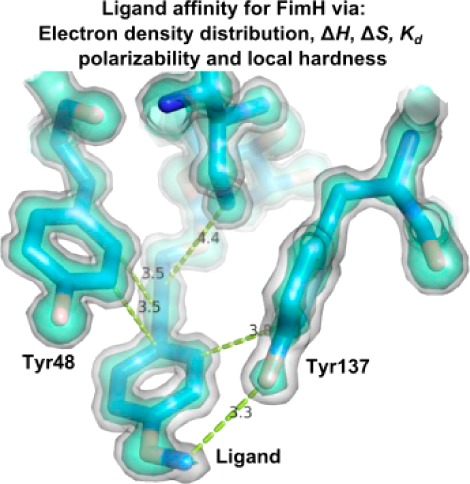
Antagonists of the FimH adhesin, a protein almost universally present at the extremity of type-1 fimbriae expressed by Escherichia coli, have been abundantly in the spotlight as alternative treatments of urinary tract infections. The antagonists function as bacterial antiadhesives through highly specific α-d-mannose binding in a charged and polar pocket at the tip of the FimH lectin domain and by the stacking of alkyl or aromatic moieties substituted on the mannose with two tyrosine residues (Tyr48 and Tyr137) at the entrance of the mannose-binding pocket. Using high-resolution crystal data, interaction energies are calculated for the different observed aromatic stacking modes between the tyrosines and the antagonist. The dispersion component of the interaction energy correlates with the observed electron density. The quantum chemical reactivity descriptors local hardness and polarizability were successfully validated as prediction tools for ligand affinity in the tyrosine gate of FimH and therefore have potential for rapid drug screening.
Keywords: FimH, stacking, local hardness, polarizability, dispersion energy, electron distribution
The receptor-binding pockets in glycan-binding proteins often include tyrosine (and tryptophane) residues with their aromatic side chains stacking against the apolar face of the ligand’s pyranoside ring.1 Such strong stacking interactions also clamp oligomannose-3 onto the hydrophobic ridge of FimH.2 A setting of two tyrosines (Tyr48 and Tyr137) backed up by an isoleucine (Ile52) gives access to the mannose-binding pocket in FimH in a way that has previously been referred to as the tyrosine gate.3 The introduction of alkyl and aryl moieties in the aglycon of synthetic α-d-mannopyranosides can mimic the stacking interactions observed of oligomannoside-3 in the tyrosine gate,2 giving important leads to construct antiadhesives that surpass the potency of known FimH inhibitors. The capability of the hydrophobic substituents on the mannose to adopt surface complementarity to the tyrosine gate has previously been indicated as an important factor in binding affinity.4 The two crystal structures of the FimH lectin domain in complex with the lead compound butyl α-d-mannoside3 demonstrated that the tyrosine gate can on its turn adapt and adopt different conformations, through tweaking the position of the Tyr48 side chain, resulting in open and closed gate conformations.4
In this work, the cocrystal structures FimH-2OH and FimH-1OMe (See Table 1 for ligand identification) are introduced. They present the closed tyrosine gate, as defined by a reorientation of the Tyr48 side chain upon ligand binding. Together with previously obtained structures of the FimH–ligand complexes with open and half-open tyrosine gates,4 they are input for quantum mechanical calculations of the interaction energies.
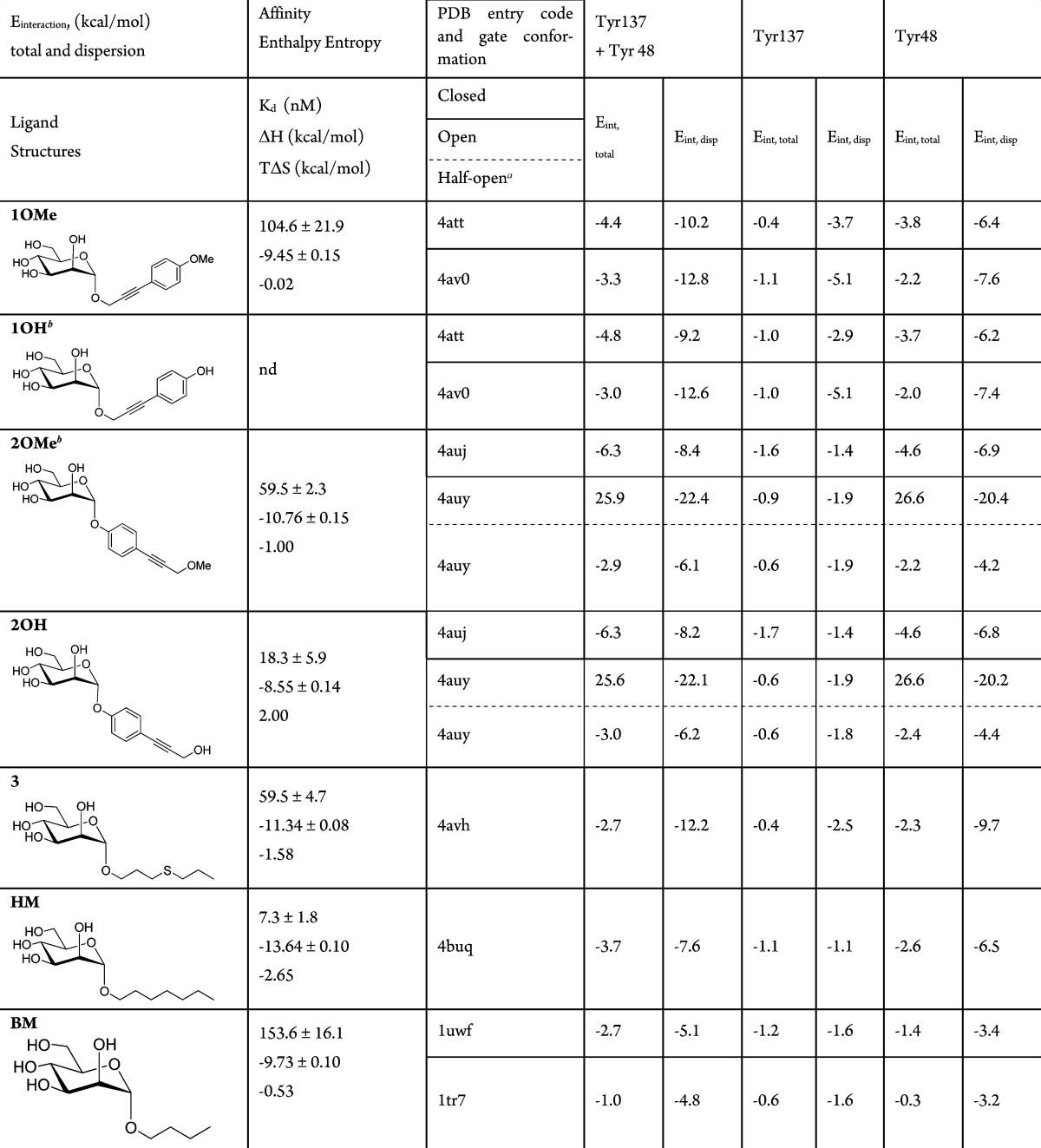
Table 1. Calculated Interaction Energies (Eint,total) and Their Dispersion (Eint,disp) Components (Eint,disp = Eint,total – Eint,HF) in kcal/mol between Ligand and Tyr137 and Tyr48 in FimH, Based on the Crystal Structures.
Half-open orientation of Tyr48 in crystals of ligand-free FimH soaked with 2OH (entry 4auy).
No crystal structure available, calculations were performed on the structures in complex with 1OMe by substituting the terminal OMe group by OH and on the structures in complex with 2OH by substituting the terminal OH group by OMe.
Details on the crystallization, data collection, and refinement of the FimH-2OH and FimH-1OMe cocrystal structures can be found in Supporting Information and Table S1. The two ligands 2OH and 1OMe have an inverted position of alkyne and phenol/phenyl methoxy groups after the α-d-mannose (Table 1). The ligand 2OH (Figure 1C) is the monovalent equivalent of a tetrameric arrangement of 2OH on pentaerythritol tetra-ester scaffold, that enabled subnanomolar affinity for the FimH E. coli adhesin.5 In the cocrystal structures, the tyrosine side chain of residue 48 closes the gate (Figure 1A,C). Tyr48 is packed in a tilted parallel position with the ligand, as observed before in cocrystal structures of FimH.3,4,6 Tyr137 is stacked in a shifted, orthogonal T-shape position with the phenyl group. The Tyr48 side chain changes significantly its orientation in the closed (Figure 1A,C) versus the open (Figure 1B,D) tyrosine gate in previously obtained structures with compounds soaked into the crystals;4 however, the ligands stay in nearly the same position.
Figure 1.
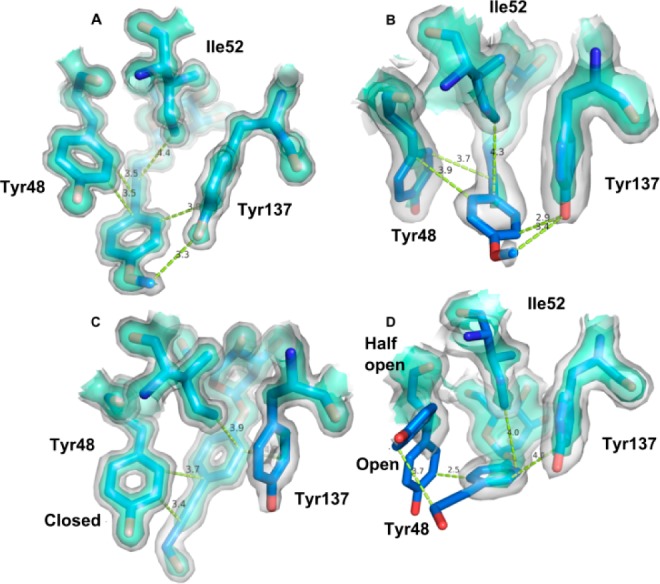
Iso-surface presentations at two levels (1σ(2Fo-Fc), white, and 2σ(2Fo-Fc), green cyan) of the electron density distribution. Residues are shown in ball-and-stick model. Distances in Å are displayed as dashed, lime-colored, lines. (A) Closed tyrosine gate in complex with α-d-mannoside O-linked to propynyl para-methoxy phenyl (1OMe, PDB entry 4att). (B) Open tyrosine gate in complex with 1OMe (PDB entry 4av0). (C) Closed tyrosine gate in complex with α-d-mannoside O-linked to p-hydroxypropargyl phenyl (2OH, PDB entry 4uj). (D) Open/half-open tyrosine gate for Tyr48, Ile52, and Tyr137 in complex with 2OH (PDB entry 4auy).
In the FimH–1OMe cocrystal structure, the phenyl ring and the conjugated bond between the phenyl ring and the propargyl group of the aglycon create apolar contacts with both tyrosines of the gate (Figure 1A). In the FimH–2OH complex (Figure 1C), the Cβ–Cγ bond of Tyr48 makes hydrophobic interactions with the phenyl moiety of ligand 2OH, with which Tyr137 stacks in a shifted T-shape orientation. The Tyr48 aromatic side chain stacks with the alkyne. The polar hydroxyl group of 2OH is in contact with the solvent and forms a hydrogen bond with a water molecule that connects to the Tyr48 hydroxyl.
To gain insight in the measured affinities, interaction energies between various ligands and the tyrosines 137 and 48 were calculated, starting from the X-ray structures (Table 1 and Supporting Information). From the interaction energies it can be concluded that the main interaction of HM and the other antagonists in crystal structures adopting a closed or half-open gate happens through Tyr48. This is consistent with Tyr137 in a shifted T-shape position toward the ligand (see, for example, Figure 1C), while Tyr48 is in a tilted parallel position above the ligand. The interaction energy is composed of a substantial amount of favorable dispersion interaction energy,7 and without dispersion, there would be no interaction, as the Hartree–Fock interaction energies (Eint,HF = Eint,total – Eint,disp excluding dispersion) are all positive.
The ligand molecules 2OH and 1OMe are about equally dynamic in both cocrystal structures (normalized against the Wilson B-factorl Table S1, Supporting Information); however, both tyrosine residues were a factor 1.6 more dynamic in the cocrystals of the complex with ligand 2OH. Tyr137 is always more dynamic (higher temperature factors) than Tyr48, but Tyr137 is more stabilized in the 1OMe (Figure 1A) than in the 2OH complex (Figure 1C). This is congruent with the almost complete lack of electron density at the 2σ(2Fo-Fc) level in the 2OH complex. In the trigonal crystals of FimH that were soaked with 2OH (PDB entry 4auy, open conformation, Table 1), the ligand dislocates the Tyr48 side chain, leading to ambiguous locations of Tyr48, one similar to the open conformation and one named the half-open conformation.4
In the open FimH–2OH complex, Tyr48 comes into a repulsive contact with the ligand (cf., the large positive total interaction energy with Tyr48 of 25.6 kcal/mol in Table 1) by which the Tyr48 is pushed away leading to the half-open Tyr48 conformation. Here, the Tyr48 side chain is in the same orientation as in the closed gate conformer (Figure 1C) but turned about 90 degrees (Figure 1D). The dislocation of the Tyr48 side chain leads to partially closing of the gate and is a logical consequence of its positive interaction energy in the open conformation. The total interaction energy in the half-open gate is indeed again negative (Table 1).
The ligand binding between ligand 2OH and Tyr48 in its open conformation is thus extremely unfavorable. From the X-ray data, it can be seen that the electron density of Tyr48 is dispersed and that the 2OH ligand is visible at the 1σ(2Fo-Fc) level only for the phenol (Figure 1D). This is in agreement with a far larger dispersion energy (−22.1 kcal/mol in Table 1) in comparison to the open form of the FimH–1OMe complex (−12.8 kcal/mol), in which the electron density is visible for the alkyne at the 2σ(2Fo-Fc) level and still largely visible for the methoxyphenyl for 1OMe at 1σ(2Fo-Fc) electron density (Figure 1B). The diffuse electron density in FimH–2OH is also reflected in the positive entropy contribution to binding (TΔS = 2 kcal/mol), combined with an enthalpic value (ΔH = −8.55 kcal/mol) that is low compared to its affinity (Table 1).
Thermodynamic data document the situation in solution and thus in the absence of stabilizing crystal lattice contacts. The positive entropy change upon interaction confirms that the dispersion energy, Eint,disp, calculated using the structure from a crystal soaked with 2OH (Table 1 and Figure 1D), plays a most important role.7 Because of the favorable contribution of the dispersion part of the interaction energy and the possibility of Tyr48 to adopt alternative conformations, binding occurs with high affinity for ligand 2OH (Kd = 18.3 nM) and also for ligand 2OMe. This indicates that the dynamics induced in the tyrosine gate by 2OH contributes positively to binding and that the open and half-open gate conformations (Figure 1D) both take part in the interactions with FimH.
All the antagonists interact stronger with Tyr48 than with Tyr137. The aromatic ligands 1OMe, 2OMe, and 2OH have a more favorable interaction energy (Eint,total, Table 1) than the nonaromatic ligands 3 and HM, in disagreement with the affinity data. This could be due to only the ligands Tyr137 and Tyr48 used in the calculations. Although this small model performs well in most cases (Figure 2), errors might be introduced by not including Ile52, a residue that also contributes to the formation of the hydrophobic pocket (Figure 1). Moreover, solvent molecules are not taken into account in the calculations of interaction energies, but those definitively play a role.2
Figure 2.

Correlation between affinity Kd (nM) and interaction energy (Eint,total) calculated based on the closed conformation. Ligand 3 and HM do not fit into the correlation (regression coefficient calculated without these ligands).
High-level MP2 calculations are computationally very demanding and only feasible on fairly small systems, making the calculation of accurate interaction energies difficult and maybe even not feasible. Therefore, there is a need to develop accurate descriptors to reliably estimate affinity data at low computational cost. For numerous ligands, even no structural information is available, and thus, no interaction energies can be calculated. Therefore, the validity of the reactivity descriptors local hardness η(r),8,9 a local version of the chemical hardness,10 and polarizability α to assess affinity is investigated here. Using these descriptors, information on the ligand affinity might be obtained without the need of calculating the interaction energy, and thus, without the need of structural information. The dispersion and the electrostatic energy components form the major part of the noncovalent π–π interaction between aromatic rings and can be related to, respectively, the polarizability α11 and the local hardness η(r). The polarizability α gives the tendency of an electron cloud to be distorted by an electric field, caused, for example, by the stacking partner. Larger polarizability is favorable for the interaction. The local hardness is a measure of negative charge accumulation.12 A large value of η(r) creates repulsion. The positions on which local hardness is calculated are chosen to be independent from the structural information (Figure S1, Supporting Information) because we aim to validate whether reactivity descriptors based on properties of the ligand alone can give information on affinity, thus prior to the availability of structural information and even prior to synthesis. The hardness and polarizability are calculated on reference systems benzene and ethyne and on compounds 1OMe and 2OH and their respective hydroxyl (1OH) and methoxy (2OMe) analogues, and on ligands 3, HM, and BM (Table 2 and described in Supporting Information).12 The components are modeled without the mannose (Figure S2, Supporting Information).
Table 2. Polarizability α (in Bohr3) and Local Hardness η(r) (in kcal/mol) in the x and y Position of the Alkyne and Alkane and in the P and T Shape Position of Benzene (Figure S1, Supporting Information).
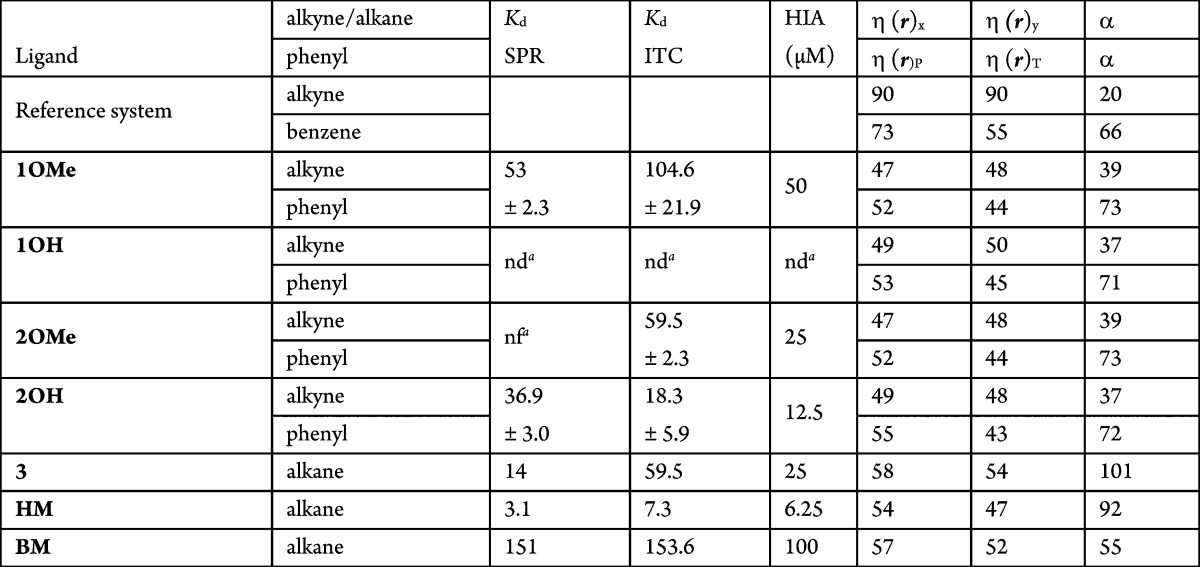
nd: not determined. nf: not fitted.
On the basis of the lowest local hardness and highest polarizability criteria, the interactions with ligands 1OMe and 2OH are overall more favorable than for the reference systems (Table 2). This might be due to the delocalization of electrons (larger polarizability) between the phenyl ring and the alkyne moiety of the ligand, resulting in less negative charge around the phenyl ring and the triple bond. As such, interaction properties of large ligand systems can differ largely from the small reference systems as benzene and ethyne. On the baisis of the polarizability, the most favorable dispersion interaction is with the phenyl group and not with the alkyne group.
Methoxy forms (1OMe and 2OMe) have lower measured affinity than hydroxylated forms (1OH and 2OH). This cannot be understood from calculated hardness and polarizability and might be explained by factors not taken into the calculations. Namely, in the crystal structures, the hydroxyl group makes multiple hydrogen bridges via water molecules to the tyrosine 137 and 48 hydroxyl groups. The ligands 1OMe and 2OMe have a very similar affinity, although the alkyne and phenyl positions are inversed.
For ligands 3, HM, and BM, the sequence of lowest local hardness (HM < BM < 3) identifies HM as the ligand with the highest affinity, in agreement with the sequence of highest measured affinity (HM < 3 < BM), but inverts the affinities for ligands 3 and BM compared to the measured ones. The sequence of the highest polarizability (3 > HM > BM) inverts the affinity of HM and 3 compared to the measured affinity (Table 2).
The reactivity descriptors predict in most cases trends that agree with affinity, indicating that they might be applied to estimate affinities for ligands prior to the availability of structural data. Here, we investigate ligands 4–9 (Table 3). Only the substituted phenyl rings are considered in the calculations (see Figure S3, Supporting Information, for the identification of the ligands). A large drop in the measured affinity has been observed between compounds 4 and 5 (Table 3). Both polarizability and local hardness are more favorable for interactions with 4 than with 5, more specifically at the nitrogen-side of 4 (Table 3). As such, the sequence of the lowest hardness and highest polarizability is consistent with the measured affinity. Although the very large increase in affinity when replacing nitrogen in 5 with sulfur in 4 (Table 3) cannot be calculated, the larger repulsion of ligand 5 compared to 4 is suggestive of aromatic stacking of their aglycon moieties with the Tyr48 side chain, in analogy with what is observed in the crystal structures (Figure 1).
Table 3. Local Hardness and Polarizability of the Substituting Phenyl Ring.
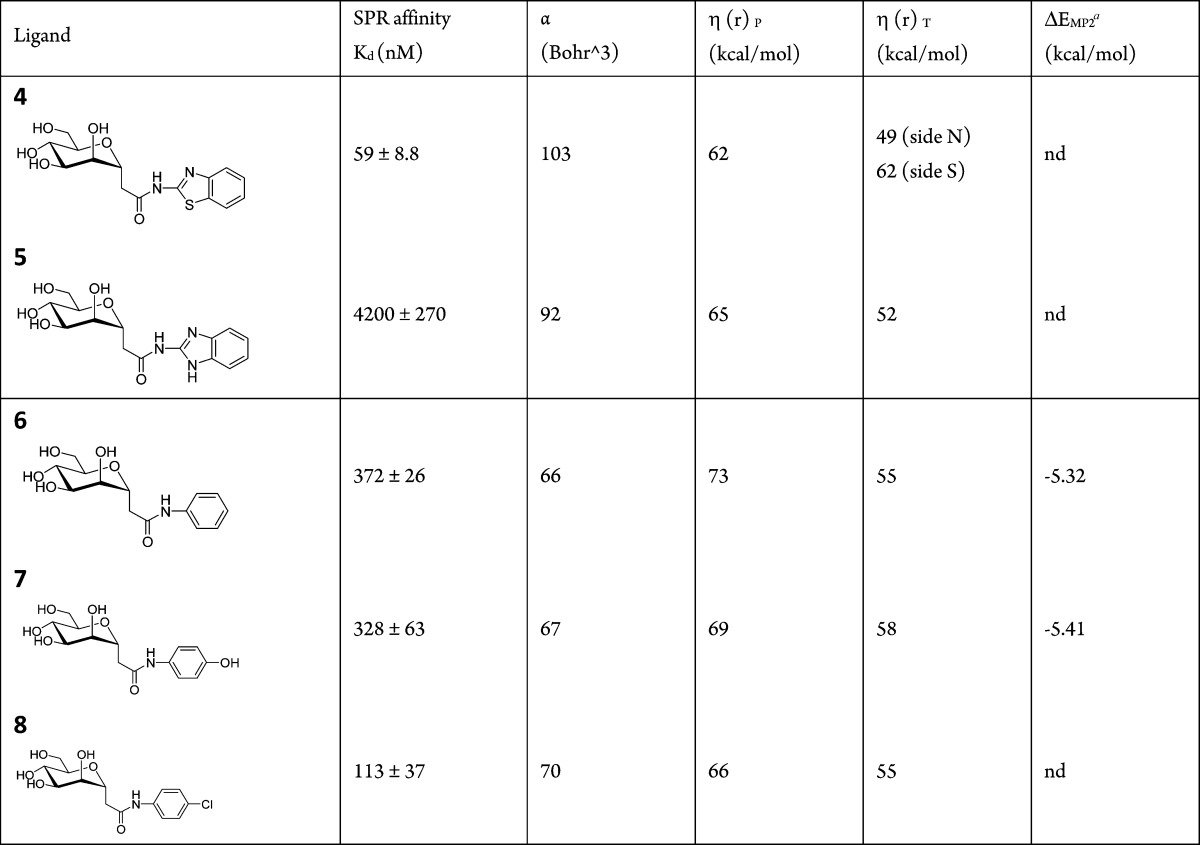
ΔEMP2: interaction energy between substituted benzene and cytosine from.12
For ligands 6 (no substitution present on the phenyl), 7 (hydroxyl in para position), and 8 (chloride halogen in para position) (Table 3 and Figure S3, Supporting Information), the sequence of the lowest hardness calculated in parallel position (−Cl < −OH < −H) and the highest polarizability (−Cl > −OH > −H) follow the sequence of the affinity (−Cl > −OH > −H); thus, the calculated hardness and polarizability prove to be useful as predictors.
In conclusion, the calculation of the interaction energies shows the central role of dispersion interactions to ensure binding. The local hardness and the polarizability can be valuable tools for rapid ligand screening prior to synthesis, when ligand series are compared or when no structural information is yet available. Further research is necessary to fine-tune the affinity prediction capacities of polarizability and local hardness by designing improved calculation protocols.
Acknowledgments
The crystal data have been collected at ProximaI of SOLEIL in Saint-Aubin, France.
Supporting Information Available
Calculation details, minimal structures used for calculation of polarizability and local hardness, and crystal data collection and refinement parameters. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by a bilateral grant between Fund for Scientific Research-Flanders (FWO) G.A060.10N-MDEIE Québec, to J.B., L.W., and R.R., and by an FWO postdoctoral fellowship to G.R. R.R. is grateful to the MDEIE-FQRNT (Québec) and NSERC for financial support of this work.
The authors declare no competing financial interest.
This paper posted ASAP on September 18, 2013. A correction was made in Table 2 and the revised version was reposted on September 26, 2013.
Supplementary Material
References
- Lemieux R. U. The origin of the specificity in the recognition of oligosaccharides by proteins. Chem. Soc. Rev. 1989, 18, 347–374. [Google Scholar]
- Wellens A.; Garofalo C.; Nguyen H.; Van Gerven N.; Slattegard R.; Hernalsteens J. P.; Wyns L.; Oscarson S.; De Greve H.; Hultgren S.; Bouckaert J. Intervening with urinary tract infections using anti-adhesives based on the crystal structure of the FimH-oligomannose-3 complex. PLoS One 2008, 3, e2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert J.; Berglund J.; Schembri M.; De Genst E.; Cools L.; Wuhrer M.; Hung C. S.; Pinkner J.; Slattegard R.; Zavialov A.; Choudhury D.; Langermann S.; Hultgren S. J.; Wyns L.; Klemm P.; Oscarson S.; Knight S. D.; De Greve H. Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesin. Mol. Microbiol. 2005, 55, 441–455. [DOI] [PubMed] [Google Scholar]
- Wellens A.; Lahmann M.; Touaibia M.; Vaucher J.; Oscarson S.; Roy R.; Remaut H.; Bouckaert J. The tyrosine gate as a potential entropic lever in the receptor-binding site of the bacterial adhesin FimH. Biochemistry 2012, 51, 4790–4799. [DOI] [PubMed] [Google Scholar]
- Touaibia M.; Wellens A.; Shiao T. C.; Wang Q.; Sirois S.; Bouckaert J.; Roy R. Mannosylated G(0) dendrimers with nanomolar affinities to Escherichia coli FimH. ChemMedChem 2007, 2, 1190–1201. [DOI] [PubMed] [Google Scholar]
- Han Z.; Pinkner J. S.; Ford B.; Obermann R.; Nolan W.; Wildman S. A.; Hobbs D.; Ellenberger T.; Cusumano C. K.; Hultgren S. J.; Janetka J. W. Structure-based drug design and optimization of mannoside bacterial FimH antagonists. J. Med. Chem. 2010, 53, 4779–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asensio J. L.; Arda A.; Canada F. J.; Jimenez-Barbero J. Carbohydrate–aromatic interactions. Acc. Chem. Res. 2013, 46, 946–954. [DOI] [PubMed] [Google Scholar]
- Chattarai P. K.; Roy D. R.; Geerlings P.; Torrent-Succarrat M. Local hardness: a critical account. Theor. Chem. Acc. 2007, 118, 923. [Google Scholar]
- Bercowitz M.; Parr R. G. On the concept of local hardness in chemistry. J. Am. Chem. Soc. 1985, 107, 6811. [Google Scholar]
- Parr R. G.; Pearson R. G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar]
- Buckingham A. D. Permanent and induced molecular moments and long-range intermolecular forces. Adv. Chem. Phys. 1967, 12, 107. [Google Scholar]
- Mignon P.; Loverix S.; Steyaert J.; Geerlings P. Influence of the pi–pi interaction on the hydrogen bonding capacity of stacked DNA/RNA bases. Nucleic Acids Res. 2005, 33, 1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.