

Abstract

We report the design and synthesis of a series of novel DGAT1 inhibitors in the benzimidazole class with a pyridyl-oxy-cyclohexanecarboxylic acid moiety. In particular, compound 11A is a potent DGAT1 inhibitor with excellent selectivity against ACAT1. Compound 11A significantly reduces triglyceride excursion in lipid tolerance tests (LTT) in both mice and dogs at low plasma exposure. An in vivo study in mice with des-fluoro analogue 10A indicates that this series of compounds appears to distribute in intestine preferentially over plasma. The propensity to target intestine over plasma could be advantageous in reducing potential side effects since lower circulating levels of drug are required for efficacy. However, in the preclinical species, compound 11A undergoes cis/trans epimerization in vivo, which could complicate further development due to the presence of an active metabolite.
Keywords: DGAT1, inhibitor, benzimidazole, ACAT1, cyclohexanecarboxylic acid, lipid tolerance test, epimerization, metabolite
Type II diabetes mellitus (T2DM) and obesity, two interlinked disease conditions, have emerged as the major threats to human health globally.1,2 In 2008, World Health Organization estimated the number of overweight and obese adults worldwide to be 1 billion and 500 million, respectively. In 2009–2010, more than one-third of the American population was classified as obese.3 Meanwhile, a recent estimate indicates that there are about 371 million people worldwide living with diabetes.4 Current drugs available to treat diabetes and obesity have limitations in terms of long-term efficacy and/or side effects.5−7 The unmet need prompts significant research efforts in this area.
Inhibition of DGAT1 (diglyceride acyltransferase 1) has emerged as a potential mechanism for the treatment of T2DM and obesity.8,9 The DGAT family (diglyce diglyceride acyltransferase 1 and 2) catalyzes the formation of triglyceride (TG) from diacylglycerol and acyl-CoA, the terminal and committed step in TG synthesis.10 DGAT1 shares only limited sequence homology with DGAT2, the other known isoform.11 In contrast, DGAT1 has more sequence homology to acyl CoA:cholesterol acyltransferase (ACAT1 and ACAT2). ACAT plays an important role in cholesterol homeostasis.12 The strong interest in DGAT1 started after the reports on DGAT1 knockout mouse phenotyping studies. DGAT1 knockout mice were shown to be viable and resistant to diet-induced obesity.13 Furthermore, these mice appeared to have increased sensitivity to insulin and leptin.14 This compelling data set has inspired major efforts in identifying small molecule DGAT1 inhibitors for potential treatment of diabetes and obesity (Figure 1).15−20 A number of drug candidates have been advanced into clinical trials. Recently, we reported that small molecule DGAT1 inhibitors markedly alter incretin peptide release following oral lipid challenge.21 Additionally, combination of DGAT1 inhibition with dipeptidyl-peptidase-4 (DPP-4) inhibition led to further enhancements in active GLP-1 in mice and dogs, suggesting potential combinability of DGAT1 inhibitors and DPP-4 inhibitors for treatment of metabolic diseases.22
Figure 1.

Structures of selected DGAT1 inhibitors.
The DGAT1 inhibitor program at Merck was primarily built upon our initial discovery of a novel benzimidazole class of compounds bearing an acid moiety at the terminus of the structure (Figure 2).23 Compounds 1 and 2 demonstrated potent inhibition against both human and mouse isoforms of DGAT1.24 However, both compounds had limited selectivity against ACAT1.25 We decided to explore analogues incorporating a pyridyl–pyridyl–ether moiety. This modification was expected to render the structure more flexible due to the ether linkage. Furthermore, the addition of nitrogen and oxygen atoms could help reduce log D. We began the work focusing on the preparation of the compounds 3A, 4A, 3B, and 4B since Cl and CF3 substitutions were shown to be favorable in the series of 1 and 2. Cyclohexanecarboxylic acid was selected as the terminal moiety since the corresponding starting materials were readily available.26
Figure 2.

Design of benzimidazole acid class with a pyridyl–pyridyl–ether moiety.
The initial method for the synthesis of this series of compounds was exemplified by the preparation of compound 3A (Scheme 1). Bromide 6A (cis) was prepared by Mitsunobu reaction of commercially available ethyl 4-hydroxycyclohexanecarboxylate (a cis and trans mixture) and 5-bromo-2-hydroxypyridine followed by SFC to separate cis and trans isomers. Next, bromide 6A was converted into pinacol boronate 7A, which underwent a Suzuki coupling reaction with 6-bromonicotinaldehyde to furnish aldehyde 8A. Oxidative condensation of 8A with 4-(trifluoromethyl)benzene-1,2-diamine formed a benzimidazole ring.27 Finally, the ester group was hydrolyzed to afford acid compound 3A. Accordingly, other compounds (3B, 4A, and 4B) were synthesized using the corresponding cis or trans isomer and the substituted benzene-1,2-diamine.
Scheme 1. Synthesis of Compound 3A.

Reagents and conditions: (a) 1. PPh3, 5-bromo-2-hydroxypyridine, DIAD, THF, 55 °C; 2. SFC (ChiralPak AD-H), first peak; (b) bis(pinacolato)diboron, KOAc, PdCl2(dppf), dioxane, 80 °C; (c) 6-bromonicotinaldehyde, Na2CO3, PdCl2(dppf), DMF–water, 80 °C; (d) 1. potassium peroxymonosulfate, 4-(trifluoromethyl)benzene-1,2-diamine, DMF–water; 2. LiOH, THF–water.
The profiles of compounds 3A/B and 4A/B are summarized in Table 1. All four compounds exhibit potent inhibition on both human and mouse DGAT1, with potencies comparable to compounds 1 and 2. These analogues also have reduced log D (HPLC) values relative to compounds 1 and 2. In addition to the excellent in vitro potency on DGAT1, all four compounds reduce triglyceride excursion in mouse LTT (lipid tolerance test).28 In particular, compound 4A demonstrates extraordinary efficacy in mouse LTT. It also gives the best selectivity (×1680) against ACAT1, judging by the ratio IC50s of ACAT1 against DGAT1 in human. Compound 4A was further evaluated for PK in rat (Table 2). It was shown to have low plasma clearance, reasonable half-life, and good plasma exposure after oral dosing.
Table 1. Profiles of Compounds 3A, 3B, 4A, and 4Ba.
| compd | human DGAT1 IC50 (nM) | mouse DGAT1 IC50 (nM) | log D HPLC | human ACAT1 IC50 (nM) | ratio of IC50 human ACAT1 vs DGAT1 | mouse LTT triglyceride reduction @ 18 h |
|---|---|---|---|---|---|---|
| 3A | 1.7 | 2.2 | 1.62 | 1093 | 643 | –84% |
| 3B | 3.0 | 4.4 | 1.72 | 1232 | 410 | –84% |
| 4A | 2.1 | 3.7 | 1.42 | 3528 | 1680 | –144% |
| 4B | 2.0 | 4.2 | 1.43 | 1344 | 672 | –83% |
Compounds were dosed in 0.5% methylcellulose at 10 mg/kg p.o. as a suspension.
Table 2. Pharmacokinetic Data of 4A in Rata.
| PK parameters | rat |
|---|---|
| F (%) | 19 |
| Cl (mL min–1 kg–1) | 3.0 |
| Vdss (L kg–1) | 0.34 |
| t1/2 (h) | 3.7 |
| AUCn (μM·h/(mg/kg)) | 2.4 |
Compound dosed in Sprague–Dawley rats as a solution in EtOH/PEG400/water (10:50:40) at 1 mg/kg, iv, and 2 mg/kg, p.o.
Given the encouraging profiles for 3A/B and 4A/B, we continued with a SAR study to assess the effect of the substitution pattern on the benzimidazole ring in both the cis and trans series in a library fashion. Scheme 2 details the chemistry for the cis series. We decided to prepare a common intermediate, which would be converted to the final analogues with minimal manipulations. For this purpose, intermediate 9A was chosen as the common intermediate instead of 8A. After surveying a few reaction conditions, we identified a good condition for hydrolyzing ester 8A to 9A without destroying the aldehyde group.29 Under the standard oxidative condensation condition, 9A reacted with a variety of diamines to give the required final compounds. With this protocol, hydrolysis of the ester group was carried out once during the preparation of common intermediate 9A rather than every time for each compound. The compounds in the trans series were prepared in a similar manner.
Scheme 2. Exploration of Substitutions on Benzimidazole Ring.

Reagents and conditions: (a) K2CO3, MeOH–water, 80 °C, 1.5 h; (b) potassium peroxymonosulfate, substituted diamine, 3% HOAc in DMF, 100 °C.
Table 3 compares the profiles for the analogues prepared from this library approach. In general, DGAT1 potency correlates well across human and mouse. Small substitutions (e.g., H and F) afford the best potency (compounds 10–12). As an exception, CN on benzimidazole maintains excellent potency on human DGAT1 but loses potency on mouse DGAT1 (compounds 13A and 13B). OCF3 (14A) gives good DGAT1 potency but suffers from poor selectivity against ACAT1.30 The sulfone, a polar group, is not tolerated for DGAT1 potency (15A and 15B). Incorporation of an azabenzimidazole core eliminates DGAT1 potency (16A and 16B). Introduction of the OMe group helps to regain reasonable DGAT1 potency but with deterioration of ACAT1 selectivity (17A and 17B).
Table 3. Profiles of Compounds 10–17.
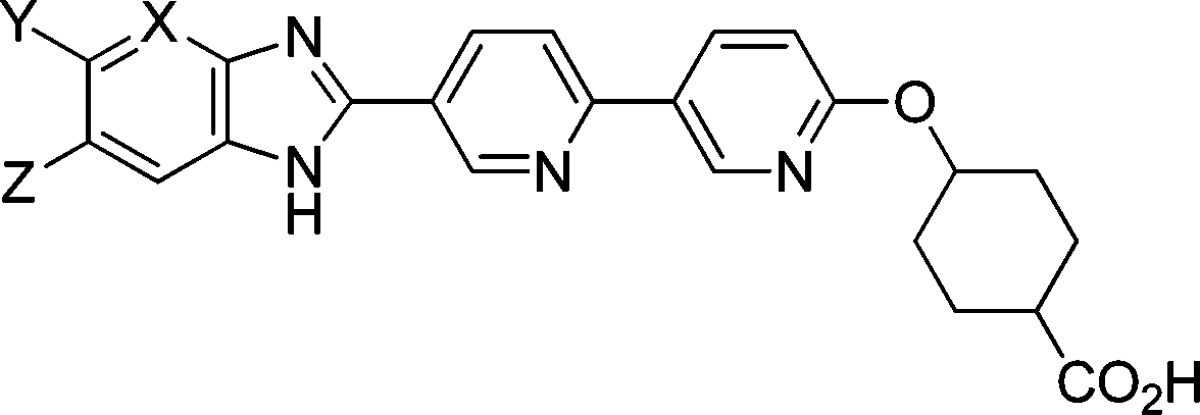
| compd | cis/trans | X | Y | Z | human DGAT1 IC50 (nM) | mouse DGAT1 IC50 (nM) | human ACAT1 IC50 (nM) |
|---|---|---|---|---|---|---|---|
| 10A | cis | CH | H | H | 4.0 | 8.1 | 8080 |
| 10B | trans | CH | H | H | 5.3 | 12 | 6065 |
| 11A | cis | CH | F | H | 2.0 | 3.4 | >10000 |
| 11B | trans | CH | F | H | 2.5 | 8.1 | >10000 |
| 12A | cis | CH | F | F | 1.4 | 3.4 | >10000 |
| 12B | trans | CH | F | F | 1.3 | 4.3 | 8440 |
| 13A | cis | CH | –CN | H | 3.2 | 36 | >10000 |
| 13B | trans | CH | –CN | H | 6.0 | 96 | >10000 |
| 14A | cis | CH | –OCF3 | H | 3.9 | 4.6 | 772 |
| 15A | cis | CH | –SO2Me | H | 101 | 358 | >10000 |
| 15B | trans | CH | –SO2Me | H | 137 | 727 | >10000 |
| 16A | cis | N | Me | H | 32 | 62 | >10000 |
| 16B | trans | N | Me | H | 54 | 161 | 8782 |
| 17A | cis | N | –OMe | H | 11 | 13 | 2475 |
| 17B | trans | N | –OMe | H | 14 | 16 | 4903 |
Overall, compound 11A gave the best profile among the analogues. As a significant advantage over 4A, compound 11A exhibited excellent selectivity against ACAT1. Furthermore, in the mouse LTT assay, compound 11A inhibited triglyceride excursion by 72% after 3 mg/kg oral dosing (with plasma trough level < 10 nM at 20 h). In a separate study, the plasma drug level of compound 11A was determined to be also low (<10 nM) at 4 h time point after oral dosing at 3 mg/kg in mice. Compound 11B, the trans isomer of 11A, also showed comparable efficacy, but compound 11A had more balanced in vitro DGAT1 IC50 numbers across human and mouse. The difluoro analogue, compound 12A, also gave a similar profile, but it has slightly higher log D (1.28) than 11A (1.02). Therefore, compound 11A was chosen for further evaluation.
To scale up compound 11A for additional profiling, we modified the chemistry (Scheme 3). The modified synthesis was more convergent and provided the material needed to support further studies. In addition, compound 10A was also scaled up according to a procedure similar to Scheme 2.
Scheme 3. Chemistry for the Scale-up of 11A.

Reagents and conditions: (a) 6-bromonicotinaldehyde, potassium peroxymonosulfate, DMF–water; (b) 1. pinacol boronate 7A, Na2CO3, PdCl2(dppf), DMF–water, 80 °C; 2. LiOH, THF–water.
In addition to the observed mouse LTT efficacy, compound 11A performed extremely well in the dog LTT assay (Figure 3).31 One hour after oral dosing of compound 11A at 3 mg/kg in 0.5% methyl cellulose, the dogs were challenged with lipid. In comparison to the vehicle group, 11A was shown to abolish lipid excursion at all time points. However, the corresponding plasma exposure at all time-points (0 to 24 h) was low (<10 nM).
Figure 3.
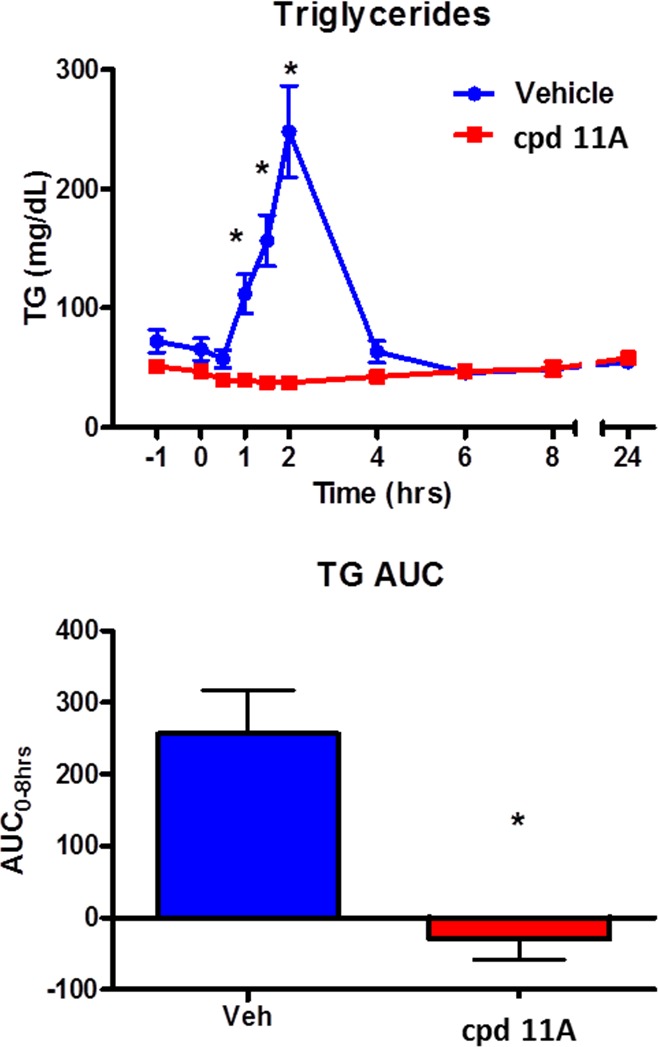
Compound 11A demonstrates excellent efficacy in dog LTT.
The apparent disconnect between the efficacy and plasma exposure of 11A could be due to the preferential tissue distribution of compound 11A in intestine, where the nutrient absorption and reassembly of triglycerides take place. This finding presented a good opportunity to develop a gut-targeting DGAT1 inhibitor, which could minimize potential side effects due to systematic plasma exposure.32
To test this hypothesis, we repeated the mouse LTT using compound 10A, the des-fluoro surrogate, since initial mouse LTT data showed 10A at 3 mg/kg dosing reduces lipid excursion by 95%, similar to compound 11A. In this study, after 3 mg/kg oral dosing, the drug levels were measured in different segments of the intestine (duodenum, jejunum, and ileum) as well as in blood at three time points (2, 5, and 25 h) (Table 4). In agreement with our hypothesis, compound 10A showed high concentration in the different segments of the intestine, much higher (>30×) than the concentration in blood at all time points.
Table 4. Concentration of 10A in Segments of Intestine and Blood after Oral Dosing at 3 mg/kg in Mouse.
| drug
level (μM) of 10A |
||||
|---|---|---|---|---|
| time point | blood | duodenum | jejunum | ileum |
| 2 h | 0.114 | 8.32 | 5.51 | 3.89 |
| 5 h | 0.051 | 8.76 | 6.01 | 1.72 |
| 25 h | 0.01 | 1.76 | 3.06 | 0.62 |
While the profiling of 11A continued, a major issue emerged when we carefully analyzed the plasma samples from the in vivo studies. In the preclinical species (rat, dog, and rhesus), after oral dosing of 11A, we detected a significant conversion of 11A to 11B by the epimerization at carbon atom attached to the carboxylic acid group. After oral dosing of 11A at 10 mg/kg in rat, the plasma exposure of metabolite 11B was about ten times the exposure of the parent 11A (Table 5). Moreover, in dog and rhesus, the plasma exposure of metabolite 11B was also comparable or higher than parent 11A. The epimerization of carboxylic acid is known and was previously studied in detail in the case of ibuprofen.33,34 It has been generally accepted that the epimerization of ibuprofen occurs enzymatically through an acyl-CoA intermediate. A similar mechanism may also operate for compound 11A. Although compound 11A has many desirable attributes, the fact that the systematic exposure of active metabolite 11B is comparable or even higher than parent 11A would likely complicate the development of 11A according to a recent FDA guidance.35 Therefore, we decided to halt the further progress of 11A.
Table 5. Pharmacokinetic Data for Compound 11Aa,b,c.
| PK parameters | rat | dog | rhesus |
|---|---|---|---|
| F (%) | 2 | 2 | 3 |
| Cl (mL min–1 kg–1) | 31.8 | 32.3 | 12.4 |
| Vdss (L kg–1) | 2.18 | 0.63 | 0.37 |
| t1/2 (h) | 1.69 | 0.82 | 2.0 |
| oral dose (mg/kg) | 10 | 2 | 2 |
| Cmax (μM) | 0.06 | 0.06 | 0.01 |
| Tmax (h) | 2.00 | 0.25 | 8.00 |
| AUC (μM·h) 11A | 0.25 | 0.05 | 0.20 |
| AUC (μM·h) metabolite 11B | 2.47 | 0.06 | 0.56 |
Compound dosed in Sprague–Dawley rats as a solution in EtOH/PEG400/water (10:50:40) at 1 mg/kg, iv, and 10 mg/kg, p.o., as solid dispersion formulation.
Compound dosed in beagles as a solution in EtOH/PEG400/water (10:50:40) at 0.55 mg/kg, iv, and as solution in 0.5% methylcellulose at 2 mg/kg, p.o.
Compound dosed in rhesus monkeys as a solution in EtOH/PEG400/water (10:50:40) at 1 mg/kg, iv, and as a solution in 0.5% methylcellulose at 2 mg/kg, p.o.
As a follow-up, a pharmacokinetic study of 11B in rat indicates that 11B is also partially converted to 11A but to a lesser extent (Table 6). After oral dosing of 11B at 2 mg/kg, the plasma exposure of metabolite 11A is roughly 10% of the parent 11B, approaching the 10% threshold recommended in the FDA guidance.35 Taking together the pharmacokinetic studies of 11A and 11B, we decided to halt this series of compounds with the cyclohexanecarboxylic acid as the terminal moiety.36
Table 6. Pharmacokinetic Data for Compound 11Ba.
| PK parameters | rat |
|---|---|
| F (%) | 24 |
| Cl (mL min–1 kg–1) | 4.2 |
| Vdss (L kg–1) | 0.46 |
| t1/2 (h) | 0.86 |
| oral dose (mg/kg) | 2 |
| AUC (μM·h) 11B | 4.47 |
| AUC (μM·h) metabolite 11A | 0.48 |
Compound dosed in Sprague–Dawley rats as a solution in EtOH/PEG400/water (10:50:40) at 1 mg/kg, iv, and 2 mg/kg, p.o.
In summary, we have described the design and synthesis of a novel series of DGAT1 inhibitors in the benzimidazole class with a pyridyl-oxy-cyclohexanecarboxylic acid moiety. Compound 11A shows excellent potency on DGAT1 and excellent selectivity against ACAT1. In addition, 11A significantly reduced triglyceride excursion in LTT tests both in mice and dogs with low plasma exposures. The excellent in vivo efficacy at low plasma exposure may be due to the preferential distribution of the compound in intestine over plasma. The ability to target the intestine over plasma could be advantageous due to possibly lower risk of potential side effects. However, this series of compounds undergo epimerization in vivo, thereby generating active metabolites. Further efforts to address the epimerization issue will be disclosed in the future.
Acknowledgments
We thank Dr. Charles W. Ross III and Mr. Thomas J. Novak at Merck Research Laboratories for measuring high-resolution mass.
Supporting Information Available
Syntheses and characterization data for the new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Scully T. Diabetes in numbers. Nature 2012, 485, S2–S3. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO). Fact sheet No. 311. Obesity and overweight. Updated March 2013. Available at: http://www.who.int/mediacentre/factsheets/fs311/en/.
- Flegal K. M.; Carroll M. D.; Kit B. K.; Ogden C. L. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. J. Am. Med. Assoc. 2012, 307, 491–497. [DOI] [PubMed] [Google Scholar]
- Guariguata L. By the numbers: new estimates from the IDF Diabetes Atlas Update for 2012. Diabetes Res. Clin. Pract. 2012, 98, 524–525. [DOI] [PubMed] [Google Scholar]
- For a recent review on pharmacologic treatment for Type-2 diabetes, see:Morsink L. M.; Smits M. M.; Diamant M. Advances in pharmacologic therapies for type 2 diabetes. Curr. Atheroscler. Rep. 2013, 15, 302. [DOI] [PubMed] [Google Scholar]
- Moller D. E. Metabolic disease drug discovery: “Hitting the target” is easier said than done. Cell Metab. 2012, 15, 19–24. [DOI] [PubMed] [Google Scholar]
- Zhang Z.-Y.; Wang M.-W. Obesity, a health burden of a global nature. Acta Pharmacol. Sin. 2012, 33, 145–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A. J.; Judd A. S.; Souers A. J. Inhibitors of diacylglycerol acyltransferase: a review of 2008 patents. Exp. Opin. Ther. Patents 2010, 20, 19–29. [DOI] [PubMed] [Google Scholar]
- Birch A. M.; Buckett L. K.; Turnbull A. V. DGAT1 inhibitors as anti-obesity and anti-diabetic agents. Curr. Opin. Drug Discovery Dev. 2010, 13, 489–496. [PubMed] [Google Scholar]
- Cases S.; Smith S. J.; Zheng Y.-W.; Myers H. M.; Lear S. R.; Sande E.; Novak S.; Collins C.; Welch C. B.; Lusis A. J.; Erickson S. K.; Farese R. V. Jr. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. 1998, 95, 13018–13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases S.; Stone S. J.; Zhou P.; Yen E.; Tow B.; Lardizabal K. D.; Voelker T.; Farese R. V. Jr. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 2001, 276, 38870–38876. [DOI] [PubMed] [Google Scholar]
- Pramfalk C.; Eriksson M.; Parini P. Cholesteryl esters and ACAT. Eur. J. Lipid Sci. Technol. 2012, 114, 624–633. [Google Scholar]
- Smith S. J.; Cases S.; Jensen D. R.; Chen H. C.; Sande E.; Tow B.; Sanan D. A.; Raber J.; Eckel R. H.; Farese R. V. Jr. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking DGAT. Nat. Genet. 2000, 25, 87–90. [DOI] [PubMed] [Google Scholar]
- Chen H. C.; Smith S. J.; Ladha Z.; Jensen D. R.; Ferreira L. D.; Pulawa L. K.; McGuire J. G.; Pitas R. E.; Eckel R. H.; Farese R. V. Jr. Increased insulin and leptin sensitivity in mice lacking acyl CoA:diacylglycerol acyltransferase 1. J. Clin. Invest. 2002, 109, 1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent reports of DGAT1 inhibitors, see:Serrano-Wu M. H.; Kwak Y.; Coppola G.; Foster C.; Gilmore T.; Gong Y.; He G.; Hou Y.; Kantor A.; Li J.; Mergo W.; Nakajima K.; Neubert A.; Radetich B.; Stroup B.; Sung M.; Szklennik P.; Tichkule R.; Yang L.; Yoon T.; Zhu Y.; Wareing J.; Hosagrahara V.; Jain M.; Chatelain R.; Commerford R.; Dardik B.; Meyers D.; Hubbard B.. Discovery of a DGAT1 inhibitor with robust suppression of postprandial triglyceride levels in humans. Abstracts of Papers, 243rd ACS National Meeting & Exposition, San Diego, CA, March 25–29, 2012. [Google Scholar]
- Dow R. L.; Li J.-C.; Pence M. P.; Gibbs E. M.; LaPerle J. L.; Litchfield J.; Piotrowski D. W.; Munchhof M. J.; Manion T. B.; Zavadoski W. J.; Walker G. S.; McPherson R. K.; Tapley S.; Sugarman E.; Guzman-Perez A.; Dasilva-Jardine P. Discovery of PF-04620110, a potent, selective, and orally bioavailable inhibitor of DGAT-1. ACS Med. Chem. Lett. 2011, 2, 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh V. S.; Beno D. W.; Brodjian S.; Brune M. E.; Cullen S. C.; Dayton B. D.; Dhaon M. K.; Falls H. D.; Gao J.; Grihalde N.; Hajduk P.; Hansen T. M.; Judd A. S.; King A. J.; Klix R. C.; Larson K. J.; Lau Y. Y.; Marsh K. C.; Mittelstadt S. W.; Plata D.; Rozema M. J.; Segreti J. A.; Stoner E. J.; Voorbach M. J.; Wang X.; Xin X.; Zhao G.; Collins C. A.; Cox B. F.; Reilly R. M.; Kym P. R.; Souers A. J. Identification and preliminary characterization of a potent, safe, and orally efficacious inhibitor of acyl-CoA:diacylglycerol acyltransferase 1. J. Med. Chem. 2012, 55, 1751–1757. [DOI] [PubMed] [Google Scholar]
- Barlind J. G.; Bauer U. A.; Birch A. M.; Birtles S.; Buckett L. K.; Butlin R. J.; Davies R. D. M.; Eriksson J. W.; Hammond C. D.; Hovland R.; Johannesson P.; Johansson M. J.; Kemmitt P. D.; Lindmark B. T.; Morentin G. P.; Noesk T. A.; Nordin A.; O’Donnell C. J.; Petersson A. U.; Redzic A.; Turnbull A. V.; Vinblad J. Design and optimization of pyrazinecarboxamide-based inhibitors of diacylglycerol acyltransferase 1 (DGAT1) leading to a clinical candidate dimethylpyrazinecarboxamide phenylcyclohexylacetic acid (AZD7687). J. Med. Chem. 2012, 55, 10610–10629. [DOI] [PubMed] [Google Scholar]
- Waring M. J.; Birch A. M.; Birtles S.; Buckett L. K.; Butlin R. J.; Campbell L.; Gutierrez P. M.; Kemmitt P. D.; Leach A. G.; MacFaul P. A.; O’Donnell C.; Turnbull A. V. Optimisation of biphenyl acetic acid inhibitors of diacylglycerol acetyl transferase 1: the discovery of AZD2353. Med. Chem. Commun. 2013, 4, 159–164. [Google Scholar]
- Goldberg F. W.; Birch A. M.; Leach A. G.; Groombridge S. D.; Snelson W. L.; Gutierrez P. M.; Hammond C. D.; Birtles S.; Buckett L. K. Discovery and optimization of efficacious neutral 4-amino-6-biphenyl-7,8-dihydropyrimido[5,4-f][1,4]oxazepin-5-one diacylglycerol acyl transferase-1 (DGAT1) inhibitors. Med. Chem. Commun. 2013, 4, 165–174. [Google Scholar]
- Liu J. Pharmacological inhibition of diacylglycerol acyltransferase 1 reduces body weight and modulates gut peptide release: potential insight into mechanism of action. Obesity 2013, 10.1002/oby.20193. [DOI] [PubMed] [Google Scholar]
- Lin H. V.; Chen D.; Shen Z.; Zhu L.; Ouyang X.; Vongs A.; Kan Y.; Levorse J. M.; Kowalik E. J.; Szeto D. M.; Yao X.; Xiao J.; Chen S.; Liu J.; Garcia-Calvo M.; Shin M. K.; Pinto S. Diacylglycerol acyltransferase-1 (DGAT1) inhibition perturbs postprandial gut hormone release. PLoS One 2013, 8, e54480. 10.1371/journal.pone.0054480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This structure class was a hybrid of the known acid series and a HTS (high throughput screening) hit containing a benzimidazole moiety. Liu J.Discovery of 2-biarylbenzimidazoles as novel DGAT1 inhibitors. Poster presented at 33rd National Medicinal Chemistry Symposium, Tucson, AZ, May 20–13, 2012
- For a description of the protocols for enzyme inhibition assays, see ref (21).
- Inhibition of ACAT has been shown to cause adverse changes in preclinical species, see:Floettmann J. E.; Buckett L. K.; Turnbull A. V.; Smith T.; Hallberg C.; Birch A.; Lees D.; Jones H. B. ACAT-selective and nonselective DGAT1 inhibition: Adrenocortical effects–a cross-species comparison. Toxicol. Pathol. 2013, 10.1177/0192623313477753. [DOI] [PubMed] [Google Scholar]
- After our work in the series was completed, an interesting report appeared recently in the literature describing DGAT1 inhibitors in the oxadiazole amide series containing this pyridyl-oxy-cyclohexanecarboxylic acid side chain:Plowright A. T.; Barton P.; Stuart B.; Alan M.; Birtles S.; Buckett L. K.; Butlin R. J.; Davies R. D. M.; Ertan A.; Gutierrez P. M.; Kemmitt P. D.; Leach A. G.; Svensson P. H.; Turnbull A. V.; Waring M. J. Design and synthesis of a novel series of cyclohexyloxy-pyridyl derivatives as inhibitors of diacylglycerol acyl transferase 1. Med. Chem. Commun. 2013, 4, 151–158. [Google Scholar]
- For the reaction condition forming benzimidazole:Beaulieu P. L.; Haché B.; von Moos E. A practical Oxone®-mediated, high-throughput, solution-phase synthesis of benzimidazoles from 1,2-phenylenediamines and aldehydes and its application to preparative scale synthesis. Synthesis 2003, 11, 1683–1692. [Google Scholar]
- For a description of the mouse LTT protocol, see ref (21).
- Jones D. A.; Nongrum F. M. Intramolecular Diels–Alder additions to 2-benzopyran-3-ones; endo-selective additions and some reactions of the adducts. J. Chem. Soc., Perkin Trans. 1 1996, 705–713. [Google Scholar]
- The corresponding trans isomer was not prepared due to the predicted poor selectivity against ACAT1 based on the data for 14A.
- For a description of the dog LTT protocol, see ref (20).
- Intestine targeted DGAT1 inhibitors were recently reported:Serrano-Wu M. H.; Coppola G. M.; Gong Y.; Neubert A. D.; Chatelain R.; Clairmont K. B.; Commerford R.; Cosker T.; Daniels T.; Hou Y.; Jain M.; Juedes M.; Li L.; Mullarkey T.; Rocheford E.; Sung M. J.; Tyler A.; Yang Q.; Yoon T.; Hubbard B. K. Intestinally targeted diacylglycerol acyltransferase 1 (DGAT1) inhibitors robustly suppress postprandial triglycerides. ACS Med. Chem. Lett. 2012, 3, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanins S. M.; Adams W. J.; Kaiser D. G.; Halstead G. W.; Hosley J.; Barnes H.; Baillie T. A. Mechanistic studies on the metabolic chiral inversion of R-ibuprofen in the rat. Drug Metab. Dispos. 1991, 19, 405–10. [PubMed] [Google Scholar]
- Knihinicki R. D.; Williams K. M.; Day R. O. Chiral inversion of 2-arylpropionic acid nonsteroidal anti-inflammatory drugs. 1. In vitro studies of ibuprofen and flurbiprofen. Biochem. Pharmacol. 1989, 38, 4389–95. [DOI] [PubMed] [Google Scholar]
- Center for Drug Evaluation and Research, U.S. FDA. Guidance for Industry: Safety Testing of Drug Metabolites, February 2008. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079266.pdf (downloaded April 2013).
- The drug levels reported in Table 2 for 4A and Table 4 for 10A are likely to be the total concentrations of the cis and trans isomers combined.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.