

Abstract
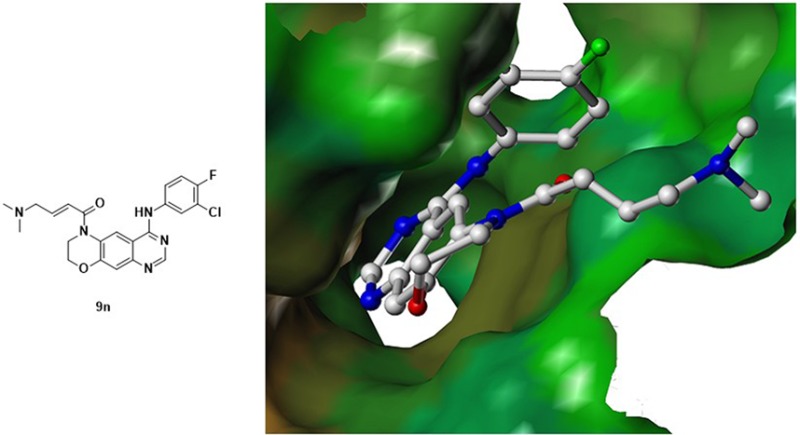
This letter describes the construction of conformationally constrained quinazoline analogues. Structure–activity relationship studies led to the identification of the lead compound 9n. Compound 9n exhibits effective in vitro activity against A431WT,overexpression and H1975[L858R/T790M] cancer cell lines but is significantly less effective against EGFR negative cancer cell lines (SW620, A549, and K562). Compound 9n was also assessed for potency in enzymatic assays and in vivo antitumor studies. The results indicated that 9n is a potent kinase inhibitor against both wild-type and T790M mutant EGFR kinase. Meanwhile, an oral administration of 9n at a dose of 200 mg/kg produced a considerable antitumor effect in a A431 xenograft model, as compared to gefitinib. A preliminary pharmacokinetic study of 9n also indicates it has good pharmacokinetic properties, and therefore, it is a good starting point for further development.
Keywords: Anticancer, kinase inhibitor, EGFR, conformationally constrained
The clinical success of a number of kinase-directed drugs indicates that kinase represent therapeutically relevant targets. The epidermal growth factor receptor (EGFR) belongs to the ErbB family of receptor tyrosine kinases (RTKs), which plays an important role in the regulation of cell growth, differentiation, and survival.1 Because of their multidimensional role in the progression of cancer, EGFR and its family members have emerged as attractive targets for anticancer therapy.2 Accordingly, targeting EGFR has been intensely pursued, with the approval of gefitinib, erlotinib, and lapatinib for use in the clinic.3−7 As is common with many therapies, challenges with respect to treatment resistance emerge over time. This situation is certainly true of EGFR inhibitor therapy. Clinical studies have demonstrated the occurrence of resistance to gefitinib with the T790M mutation accounting for 50% of the clinically observed resistant mutations.
In order to overcome the T790M mutation related drug resistance, a variety of irreversible inhibitors were developed. These compounds contain a Michael acceptor moiety designed to form a covalent bond with the conserved cysteine residue (Cys797) at the lip of the EGFR ATP binding site.8,9 So far, canertinib,10,11 neratinib,12,13 dacomitinib,14 and afatinib15 have been developed as second generation irreversible inhibitors with limited clinical efficacy. These drugs demonstrate the utility of quinazoline derivatives as an attractive scaffold for the development of EGFR inhibitors. To date, many studies have been targeted at finding new structures based on quinazolines that are potent EGFR inhibitors.16−18 All of these inhibitors share a common feature: upon the binding to EGFR, a hydrogen bond forms between the N1 atom of the compound and the backbone NH of Met769 in the hinge region.
To investigate the potential use of a novel scaffold as an EGFR ATP binding site inhibitor, a series of conformationally constrained quinazoline derivatives were synthesized (consisting of 34 structurally characterized compounds with purity >98%). (For general synthetic procedures refer to Schemes 1 and 2 and Supporting Information.) From this set of new compounds, a specific analogue was identified that inhibits the growth of both gefitinib-resistant (H1975[T790M/L858R]) and gefitinib-sensitive (A431WT,overexpression) cell lines but is not effective against EGFR-negative (A549, K562, and SW620) cells.
Scheme 1. Synthesis of Compounds 7a–e and 8a–b.

Scheme 2. Synthesis of Compounds 9a–9p.
The screening result (Table S1, Supporting Information) of the synthesized analogues 7a–7o and 8a–8b did not show obvious inhibition on the proliferation of A431 human epithelial carcinoma cells with overexpressed EGFRWT, but surprisingly, some of the synthesized analogues suppressed the enzymatic activity of EGFRWT, which might be due to the complex genomic background and the strong binding of EGFRWT with ATP.19 For instance, 7b inhibited the enzymatic activities of EGFRWT with an IC50 value of 1.29 nM, but its activity on A431 cancer cells was approximately 20 000 times less potent (IC50 value was more than 30 μM). The results indicated that a further modification of the scaffold might prove to be an effective strategy for identifying more potent inhibitors. The impact of the R3 group was first studied by replacement with other acrylamide groups. Interestingly, the inhibitory potency against A431WT,overexpression and wild-type EGFR was significantly increased by over 20-fold (Table S2, Supporting Information). For instance, when the hydrogen on the R3 substitution was replaced with acrylamide, the resulting compounds 9a–9e are almost 5 times more potent than 7a–7e. However, when the substituted acrylamide was replaced with a slightly hydrophobic moiety (9f–9h), the potencies against A431WT,overexpression and wild-type EGFR kinase were almost completely abolished. Further investigation also revealed that the potency loss of 9f–9h was partially restored by introducing a N-morpholino moiety (9i–9l) at the position of the original phenyl moiety. The result suggests that the substituted acrylamide at the R3 position can be replaced with a more hydrophilic group in order to improve the potency against the A431 and wild-type EGFR kinases. This hypothesis was verified by the in vitro screening result of 9m–9p. The inhibitory activities against A431WT,overexpression and EGFRWT were improved, with IC50 values almost equal to the reference irreversible inhibitor, canertinib. We further profiled the synthesized analogues 9a–9p against NSCLC cell line H1975. The H1975 cell line bears the drug resistant mutation in EGFR[L858R/T790M] and is a typical EGFR-driven cell lines. Several compounds (8c–8d and 9n–9p) also displayed strong antiproliferative effects on gefitinib-resistant H1975 cells, with the IC50 value equal to or more potent than that of canertinib (Table S2, Supporting Information). Three closely related analogues, 9n–9p, were identified from the screen that possessed approximately equal IC50 values against A431WT,expression and H1975[L858R/T790M] as the reference inhibitors (Figure 1 and Tables S2 and S3, Supporting Information).
Figure 1.
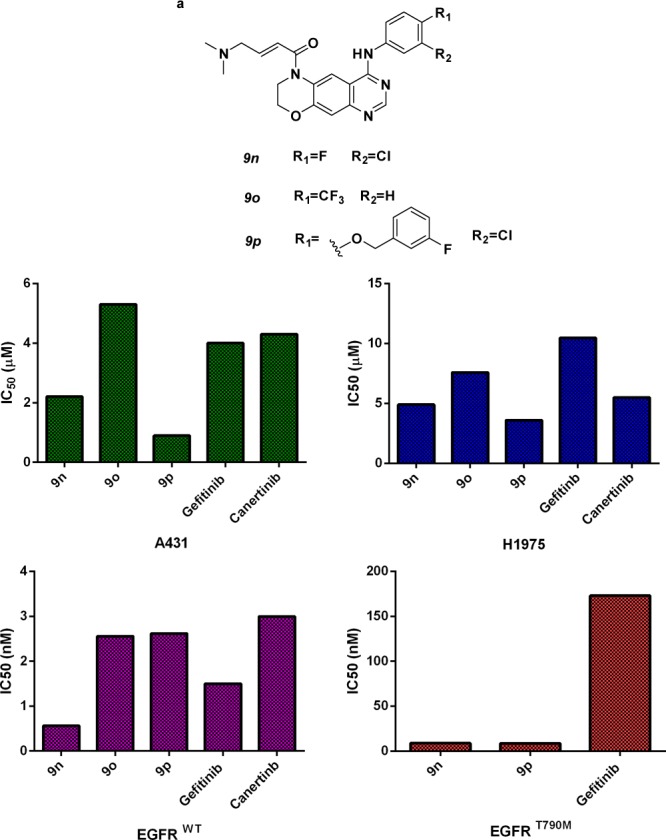
Compounds 9n–9p are novel EGFR inhibitors, suppress the growth of EGFRWT/EGFRL858R/T790M containing cancer cell lines, and inhibit the enzymatic activities of EGFRWT/EGFRT790M kinase. (a) Chemical structures of 9n–9p. IC50 values for A431 (top left), H1975 (top, right), EGFRWT (bottom, left), and EGFRT790M (bottom, right). Growth inhibitory was assessed using the MTT.
To better understand how our conformationally constrained compounds interact with the ATP-binding cleft of EGFR and how this relates to their potency for EGFRT790M, we determined a putative binding mode of one representative compound 9n within the active binding pocket of the EGFRT790M (Figure 2). The morpholine fused scaffold is located in the ATP-binding pocket. Two highly conserved hydrogen bonds are formed between the quinazoline core and the hinge region. The molecule is oriented such that 6-substituted acrylamide branch extends toward the solvent, and the 4-aniline moiety is directed into the backpocket of EGFR kinase. The docking result is shown to form a covalent bond with Cys797 as expected, with a measured distance of 2.54 Å. The prediction is consistent with observed result from Western blot studies (Figure 3). In addition, a salt bridge was formed between the carboxyl group of Asp800 and the nitrogen atom of the dimethylamino group. Furthermore, the protonated dimethylamino group might facilitate the nucleophilic attack of the sulfhydryl group of Cys797.20 The prediction is also consistent with our observations that a hydrophobic substituent (e.g., phenyl) led to a decrease in the potency in comparison with the dimethylamino group.
Figure 2.
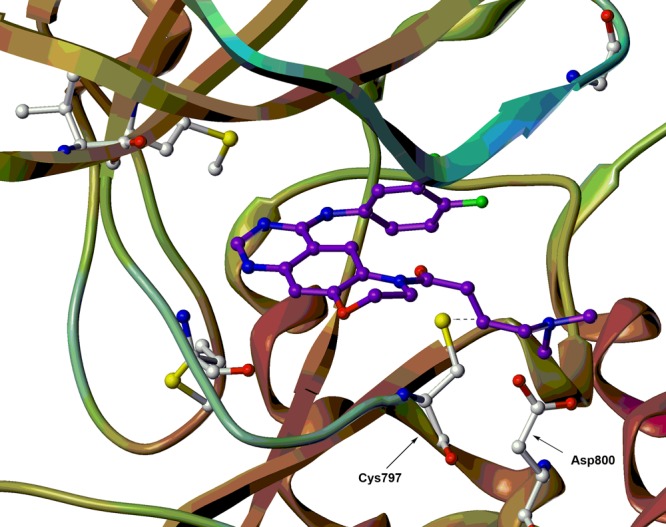
Putative binding mode of 9n within the active pocket of the EGFRT790M (PDB code: 4I24).
Figure 3.
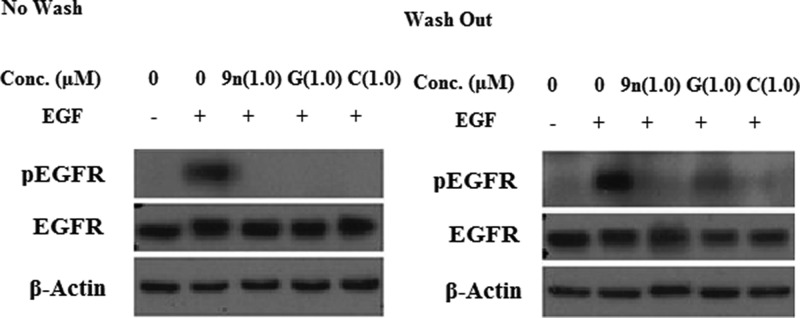
Inhibition of EGFR autophosphorylation in A431 by Western blot assay. G, gefitinib; C, canertinib.
Compound 9n and canertinib inhibit the autophosphorylation of EGFR in no wash (left) and wash out (right), while gefitinib has no effects in the wash out group. The result is the evidence of irreversible binding mode, suggesting 9n is acting the same as irreversible inhibitor canertinib.
The in vitro data shown in Figure 1 demonstrates that the new synthesized analogues 9n–9p strongly inhibit the EGFRWT kinase. Because many tissues use wild-type EGFR for normal cellular processes, the potential for toxicity from irreversible EGFR inhibitors is a concern. Therefore, the growth inhibitory of the compounds against various cancer cell lines was evaluated to control the potential toxicity. As shown in Table 1, the potent EGFR inhibitor 9n did not display obvious inhibition on the growth of A549, SW620, and K562 cancer cells, all of which have low levels of EGFR expression. The results indicate that the cytotoxic effects of 9n are minimal. Meanwhile, in the human ether-a-go-go-related gene (hERG) potassium channel patch clamp assay,219n has an IC50 >10 μM, which indicates it has a low potential for cardiac toxicity (Table S4, Supporting Information).
Table 1. In Vitro Growth Inhibitory Activities against Various Cancer Cell Lines.
| cancer type | cell line | characteristic | 9n (μM) | 9p (μM) | gefitinib (μM) |
|---|---|---|---|---|---|
| epidemal | A431 | EGFRWT,overexpression | 2.2 | 0.9 | 4.0 |
| NSCLC | H1975 | EGFRL858R/T790M | 4.9 | 3.61 | 10.48 |
| NSCLC | A549 | EGFRWT,κ-Ras mutation | >10 | >10 | >10 |
| colon | SW620 | EGFR negative | 25.4 | 6.2 | 28.20 |
| malignant myeloid | K562 | EGFR negative | >10 | >10 | >10 |
Furthermore, 9n and 9p have a good pharmacokinetic in vitro ADME profiles: the inhibition of cytochrome P450 was assessed in recombinant human cytochrome P450 isoforms (1A2, 2C9, 2D6, and 3A4), and the IC50 values were higher than 10 μM, suggesting a low potential for 9n and 9p to be involved in any drug–drug interactions. The intrinsic clearance found in liver microsomes predicts good to excellent in vivo clearance (Table S5, Supporting Information).
A pharmacokinetic study of 9n and 9p was performed in BALB/c mice. As shown in Table 2, 9n demonstrated desirable results with a half-life (9.1 h) and oral bioavailability (26.1%), while 9p displayed an unsatisfactory pharmacokinetic prolife (F = 6.42%) (Table S6, Supporting Information).
Table 2. Pharmacokinetic Parameters for 9na.
| route | AUC(0–24 h) (μM·min) | t1/2 (min) | tmax (min) | Cmax (μM) | F% |
|---|---|---|---|---|---|
| i.v. | 25.4 | 762 | 2 | 0.29 | |
| p.o. | 133 | 548 | 45 | 0.36 | 26.1 |
The pharmacokinetic parameters are obtained after a single i.v. (3 mg/kg) or oral (10 mg/kg) administration. The data are obtained from 12 mice in each treatment group.
We further determined the effectiveness of 9n in vivo by using a nude mouse xenograft model harboring EGFRWT,expression. We chose 9n for the in vivo studies because, in vitro, it is effective against EGFR expressing cancer cell lines and has a good ADME prolife. Gefitinib (200 mg/kg) was used as a reference drug to validate the models. In the A431 tumor model, the mice were treated via oral gavage once daily with either 9n or gefitinib once the tumor had grown to a volume of 50–150 mm2. Oral administration of 9n and gefitinib at 200 mg/kg/day inhibited tumor growth at 66.8% and 35.9%, respectively. No mortality or significant weight loss was observed during the treatment (Figure 4).
Figure 4.
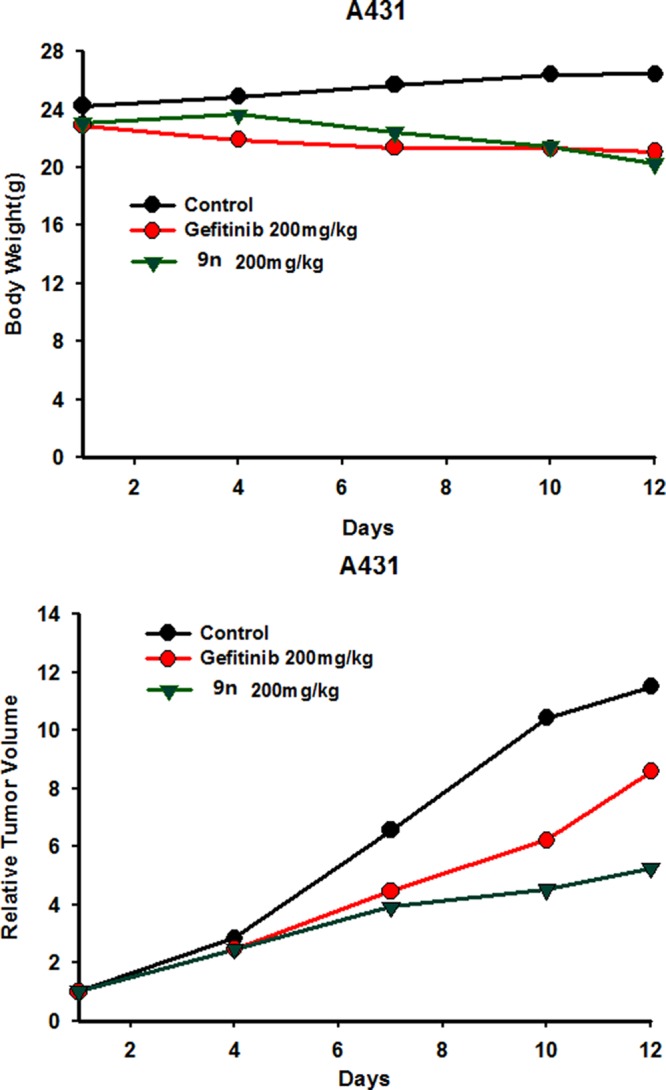
In vivo antitumor effects of compound 9n. (top) Body weight curves for the A431 xenograft model. (bottom) Tumor growth curves for the A431 xenograft model. Animals were randomized into groups (n = 6/group) and administered 9n, gefitinib, and vehicle once daily at the indicated dose level when the tumors reached the determined size, and the animal weight and tumor volume were monitored twice weekly. Data points represent the mean tumor mass (SEM of 6 mice). Relative tumor growth is calculated by mean tumor mass on day 4, 7, 10, or 12 divided by mean tumor mass on day 0. A p-value (p < 0.05) indicates a statistically significant reduction in relative tumor growth of the treated groups compared with the control group.
In summary, a series of conformationally constrained quinazoline derivatives have been designed and synthesized and shown utility as EGFR inhibitors. The most potent compounds 9n and 9p strongly inhibited the enzymatic activities of wild-type EGFR kinase as well as clinical resistant EGFRT790M mutant kinase. The kinase inhibitory efficiency of the compounds were further validated by Western Blot analysis for the activation of EGFR. Further in vitro assay demonstrated that 9n and 9p are effective against H1975 nonsmall cell lung cancer cells bearing EGFR[L858R/T790M], with potencies better than gefitinib. Compounds 9n and 9p showed minimal cytotoxicity to K562 and SW620. A hERG assay demonstrated their unlikely cardiac toxicity, indicating that these analogues might possess a high safety index. An in vivo antitumor assay demonstrates that an oral once daily dose of 9n at 200 mg/kg produces considerable tumor inhibition in the A431 xenograft model, as compared to gefitinib. The pharmacokinetic studies indicate that 9n possesses good pharmacokinetic properties. In conclusion, our studies identified a new lead compound containing a novel scaffold ideally suited for the development of therapeutically relevant EGFR inhibitor effective against EGFRT790M mutations. Further detailed studies of the mechanisms of action for 9n are currently underway.
Acknowledgments
We are grateful to the financial support and biological activity test from Shanghai Pharmaceuticals Holding Co., Ltd. We also appreciated Marc A. Giulianotti (Torrey Pines Institute for Molecular Studies, USA) for critical reading of the manuscript.
Supporting Information Available
Details of the synthesis and characterization of all compounds and intermediates together with protocols for biological experiments, some biological data, and computational methods used for molecular docking This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ J.W. and W.C. contributed equally to this work.
This research was supported by the National Natural Science Foundation of China (No. 81072516 and No. 81273356), Natural Science Foundation of Zhejiang Province (No. Z2110655), Program for Zhejiang Leading Team of S&T Innovation, and the Osteoporosis and Breast Cancer Research Center, USA.
The authors declare no competing financial interest.
Supplementary Material
References
- Olayioye M. A.; Neve R. M.; Lane H. A.; Hynes N. E. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwind A.; Fischer O. M.; Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [DOI] [PubMed] [Google Scholar]
- Adjei A. A. Epidermal growth factor receptor tyrosine kinase inhibitors in cancer therapy. Drugs Future 2001, 26, 1087–1092. [Google Scholar]
- Renhowe P. A. Inhibitors of growth factor receptor kinase-dependent signaling pathways in anticancer chemotherapy-clinical progress. Curr. Opin. Drug Discovery Dev. 2002, 5, 214–224. [PubMed] [Google Scholar]
- Ranson M.; Hammond L. A.; Ferry D.; Kris M.; Tullo A.; Murray P. I.; Miller V.; Averbuch S.; Ochs J.; Morris C.; Feyereislova A.; Swaisland H.; Rowinsky E. K. ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: results of a phase I trial. J. Clin. Oncol. 2002, 20, 2240–2250. [DOI] [PubMed] [Google Scholar]
- Ciardiello F.; Tortora G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [PubMed] [Google Scholar]
- Johnston S. R. D.; Leary A. Lapatinib: A novel EGFR/HER2 tyrosine kinase inhibitor for cancer. Drugs Today 2006, 42, 441–453. [DOI] [PubMed] [Google Scholar]
- Leproult E.; Barluenga S.; Moras D.; Wurtz J.-M.; Winssinger N. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: Application to the design of selective covalent inhibitors. J. Med. Chem. 2011, 54, 1347–1355. [DOI] [PubMed] [Google Scholar]
- Potashman M. H.; Duggan M. E. Covalent modifiers: An orthogonal approach to drug design. J. Med. Chem. 2009, 52, 1231–1246. [DOI] [PubMed] [Google Scholar]
- Smaill J. B.; Showalter H. D. H.; Zhou H.; Bridges A. J.; McNamara D. J.; Fry D. W.; Nelson J. M.; Sherwood V.; Vincent P. W.; Roberts B. J.; Elliott W. L.; Denny W. A. Tyrosine kinase inhibitors. 18. 6-Substituted 4-anilinoquinazolines and 4-anilinopyrido-[3,4-d]pyrimidine as soluble, irreversible inhibitors of the epidermal growth factor receptor. J. Med. Chem. 2001, 44, 429–440. [DOI] [PubMed] [Google Scholar]
- Smaill J. B.; Rewcastle G. W.; Loo J. A.; Greis K. D.; Chan O. H.; Reyner E. L.; Lipka E.; Showalter H. D. H.; Vincent P. W.; Elliott W. L.; Denny W. A. Tyrosine kinase inhibitors: 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazolineand 4-(phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides bearing additional solubilizing functions. J. Med. Chem. 2000, 43, 1380–1397. [DOI] [PubMed] [Google Scholar]
- Tsou H. R.; Overbeek-Klumpers E. G.; Hallett W. A.; Reich M. F.; Floyd M. B.; Johnson B. D.; Michalak R. S.; Shen R.; Shi X.; Wang Y. F.; Upeslacis J.; Wissner A. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 2005, 48, 1107–1131. [DOI] [PubMed] [Google Scholar]
- Wissner A.; Overbeek E.; Reich M. F.; Floyd M. B.; Johnson B. D.; Mamuya N.; Rosfjord E. C.; Discafani C.; Davis R.; Shi X.; Rabindran S. K.; Gruber B. C.; Ye F.; Hallett W. A.; Nilakantan R.; Shen R.; Wang Y.; Greenberger L. M.; Tsou H. Synthesis and structure-activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles: The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2). J. Med. Chem. 2003, 46, 49–63. [DOI] [PubMed] [Google Scholar]
- Engelman J. A.; Zejnullahu F.; Gale C.-M.; Lifshits E.; Gonzales A. J.; Shimamura T.; Zhao F.; Vincent P. W.; Naumov G. N.; Bradner J. E.; Althaus I. W.; Gandhi L.; Shapiro G. I.; Nelson J. M.; Heymach J. V.; Meyerson M.; Wong K.-K.; Jänne P. A. PF00299804, an irreversible Pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007, 67, 11924–31192. [DOI] [PubMed] [Google Scholar]
- Li D.; Ambrogio L.; Shimamura T.; Kubo S.; Takahashi M.; Chirieac L. R.; Padera R. F.; Shapiro G. I.; Baum A.; Himmelsbach F.; Rettig W. J.; Meyerson M.; Solca F.; Greulich H.; Wong K. K. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Lv P.; Zhang H.; Zhang H.; Hou Y.; Liu K.; Ye Y.; Zhu H. The combination of 4-anilinoquinazoline and cinnamic acid: a novel mode of binding to the epidermal growth factor receptor tyrosine kinase. Bioorg. Med. Chem. 2011, 19, 5012–5022. [DOI] [PubMed] [Google Scholar]
- Ballard P.; Barlaam B.; Bradbury R. H.; Dishington A.; Hennequin L. F.; Kickinson D. M.; Hollinsworth I. M.; Kettle J. G.; Klinowska T.; Ogilvie D. J.; Pearson S. E.; Scott J. S.; Suleman A.; Whittaker R.; Williams E. J.; Wood R.; Wright L. Neutral 5-substituted 4-anilinoquinazolines as potent, orally active inhibitors of erbB2 receptor tyrosine kinase. Bioorg. Med. Chem. Lett. 2007, 17, 6326–6329. [DOI] [PubMed] [Google Scholar]
- Barlaam B.; Ballard P.; Bradbury R. H.; Ducray R.; Germain H.; Kickinson D. M.; Hudson K.; Kettle J. G.; linowska T. K.; Magnien F.; Ogilvie D. J.; Olivier A.; Pearson S. E.; Scott J. S.; Suleman A.; Trigwell C. B.; Vautier M.; Whittaker R. D.; Wood R. A new series of neutral 5-substituted 4-anilinoquinazolines as potent, orally active inhibitors of erbB2 receptor tyrosine kinase. Bioorg. Med. Chem. Lett. 2008, 18, 674–678. [DOI] [PubMed] [Google Scholar]
- Carey K. D.; Garton A. J.; Romero M. S.; Kahler J.; Thomson S.; Ross S.; Park F.; Haley J. D.; Gibson N.; Sliwkowski M. X. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006, 66, 8163–8171. [DOI] [PubMed] [Google Scholar]
- Tsou H.-R.; Mamuya N.; Johnson B. D.; Reich M. F.; Gruber B. C.; Ye F.; Nilakantan R.; Shen R.; Discafani C.; DeBlanc R.; Davis R.; Koehn F. E.; Greenberger L. M.; Wang Y.-F.; Wissner A. 6-Substituted-4-(3-bromophenylamino)quinazolines epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor (HER-2) tyrosine kinases with enhanced antitumor activity. J. Med. Chem. 2001, 44, 2719–2734. [DOI] [PubMed] [Google Scholar]
- Diaz G. J.; Daniell K.; Leitza S. T.; Martin R. L.; Su Z.; McDermott J. S.; Cox B. F.; Gintant G. A. The [3H]-dofetilide binding assay is a predictive screening tool for hERG blockade and proarrhythmia: comparison of intact cell and membrane preparations and effects of altering [K+]o. J. Pharmacol. Toxicol. Methods 2004, 50, 187–199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.