

Abstract
In order to identify novel Alzheimer’s modifying pharmacological tools, we developed bis-tacrines bearing a peptide moiety for specific interference with surface sites of human acetylcholinesterase (hAChE) binding amyloid-beta (Aβ). Accordingly, compounds 2a–c proved to be inhibitors of hAChE catalytic and noncatalytic functions, binding the catalytic and peripheral sites, interfering with Aβ aggregation and with the Aβ self-oligomerization process (2a). Compounds 2a–c in complex with TcAChE span the gorge with the bis-tacrine system, and the peptide moieties bulge outside the gorge in proximity of the peripheral site. These moieties are likely responsible for the observed reduction of hAChE-induced Aβ aggregation since they physically hamper Aβ binding to the enzyme surface. Moreover, 2a was able to significantly interfere with Aβ self-oligomerization, while 2b,c showed improved inhibition of hAChE-induced Aβ aggregation.
Keywords: Cholinesterase inhibitors, amyloid beta-peptides, multifunctional tools, amyloid beta oligomers, Alzheimer’s disease, bivalent ligands
Convergent biochemical and genetic evidence suggest that the formation of amyloid-beta peptides (Aβ) deposits in the brain is an important seminal step in the development of Alzheimer’s disease (AD). The assembly of Aβ into a variety of oligomeric and fibrillar species is one of the causal factors of AD. Aβ oligomers have been shown to accelerate neuron cell death and are therefore thought to precipitate synaptic dysfunction. The inhibition of Aβ oligomerization could thus provide a novel approach for treating the underlying cause of AD.1 This latter aspect could be combined to inhibition of cholinesterases (ChE) catalytic activity and to interference with acetylcholinesterase (AChE) accelerated Aβ aggregation in a single disease-modifying anti-Alzheimer’s drug (DMAAD). The multifactorial nature of AD indeed supports a therapeutic approach based on multitarget directed ligands.2 Currently available therapies for AD are only symptomatic,3 and inhibition of AChE and butyrylcholinesterase (BuChE) is to date the most established therapeutic approach.4−6 AChE interacts with Aβ by a mechanism involving its peripheral anionic site (PAS), and it was proposed that AChE may accelerate the deposition of Aβ into fibrils7 (noncatalytic function of AChE).8−11 Further studies indicate that BuChE also colocalizes with Aβ in senile plaques, and may play a role in plaques maturation.12 While deposition of Aβ plaques is the hallmark of the disease, the neurotoxicity of Aβ oligomers was shown to be stronger than that of the fibrils.13,14 Therefore, innovative DMAAD should also possess inhibition properties against Aβ self-association. On these bases, we have developed a new set of multifunctional pharmacological tools that inhibit Aβ self-association and oligomerization, the enzymatic activity of ChEs, and (to a moderate extent) the AChE-induced Aβ aggregation (2a–c, Figure 1). Tacrine-based bisfunctional ligands inhibit AChE hydrolytic activity,15 and in some cases, they interfere with its noncatalytic functions (binding to Aβ), by interacting with AChE PAS (W279, Torpedo californica AChE (TcAChE) numbering).10,15−18 Such inhibitors span the active-site gorge, and the nature of the linker affects the affinity for AChE and BuChE (dual or triple sites inhibitors 1a–c, Figure 1).15 In order to develop innovative multifunctional pharmacological tools, based on our previous experience, we synthesized compounds 2a–c (Figure 1) by combining a bis-tacrine scaffold (for achieving hChEs inhibition) with a hydrophobic peptidomimetic sequence to interfere with the putative surface binding region of Aβ around W279, thus interfering with AChE-induced Aβ aggregation and Aβ self-association.13
Figure 1.
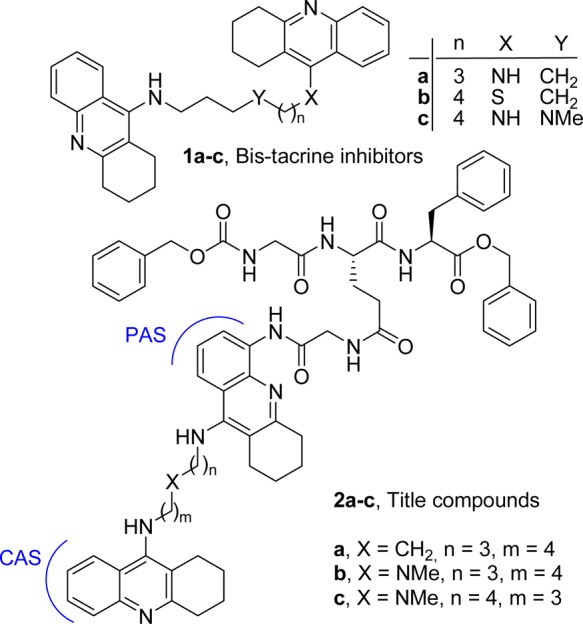
Reference and title compounds.
Molecular modeling studies (Figure 2) and biological studies (Table 1 and Figure 3) confirmed our working hypothesis. Particularly, molecular docking procedure within TcAChE provided a clear-cut vision of the interaction pattern of 2a–c with TcAChE catalytic site (CAS), PAS, and its surrounding surface area, allowing rationalization of the experimental data. The synthesis of compounds 2a–c is reported in Scheme 1 of the Main Text and Scheme 1SI of the Supporting Information; discussion of the procedure is given in the Supporting Information. Biological data are reported in Table 1. The inhibition tests performed on hChEs revealed that 2a (as well as precursors 6 and 7) is a potent reversible inhibitor of hChEs (KihAChE = 1.9 nM, KihBuChE = 40.9 nM). Although both ChEs colocalize with Aβ plaques, 2a was only tested against hAChE-induced Aβ1–40 aggregation. It also exhibited potent inhibition of Aβ1–42 spontaneous aggregation (81% at 5 μM). The potency of 2a against hAChE-induced Aβ aggregation was improved using the tether of 1c, one of the most potent inhibitors of hAChE known to date.15 The selection was based on its specific interaction with the three identified recognition sites of the hAChE gorge (observed by docking studies using hAChE),15 which pushes the PAS oriented tacrine moiety to establish a triple π–π stacking with W286 and Y72 thus inducing a rotation of W286 (hAChE numbering).15 Accordingly 2b,c demonstrated higher ability to inhibit hAChE Aβ aggregation while maintaining nM inhibition potency for hChEs.
Figure 2.

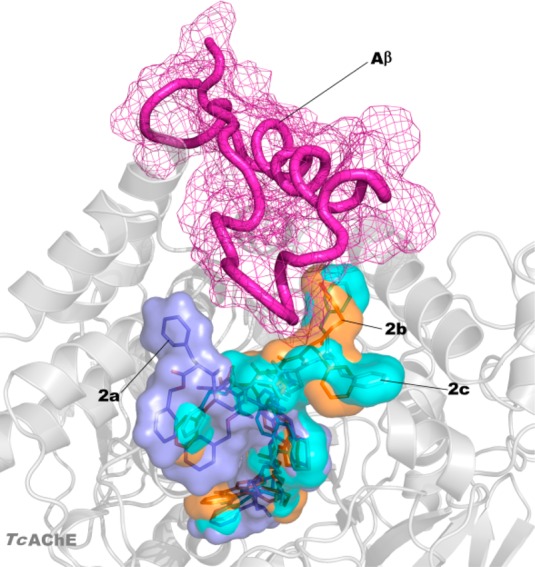
Docked poses of compounds 2a–c into the TcAChE binding site (key residues are represented by lines) obtained using IFD protocol: (A) 2a (blue sticks, blue surface for protein, glide XP score: −19.603 kcal/mol); (B) 2b (orange sticks, yellow surface for protein, glide XP score: −20.561 kcal/mol); (C) 2c (cyan sticks, green surface for protein, glide XP score: −21.143 kcal/mol). H-bonds were reported as gray dotted lines. Nonpolar hydrogens were omitted for clarity. The picture was generated by PyMOL.
Table 1. Inhibition of hChEs, Aβ1-42 Spontaneous, and hAChE-Induced Aβ1-40 Aggregation.
| compd | Ki (nM) hAChEa | Ki (nM) hBuChEa | hAChE/hBuChE | Aβ1–42 aggreg at 5 μM (%)b | Aβ1–40hAChE-induced aggreg (%)b |
|---|---|---|---|---|---|
| 1a | 8.4c | 68e | |||
| 1b | 28.0 | 1.65 | 16.9 | NT | NT |
| 1c | 0.012 | 0.82 | 0.01 | NT | 50 |
| 6 | 0.78 | 0.06 | 13 | NT | NT |
| 7 | 0.23 | 8.26 | 0.028 | NT | NT |
| 2a | 1.92 | 49.8 | 0.038 | 81 | 26 |
| 2b | 5.02 | 61.77 | 0.081 | NT | 42 |
| 2c | 1.48 | 21.26 | 0.070 | 51d | 42f |
SD were within 10% of the mean.
SEM were within 10% of the mean.
Ref (19).
μM IC50 value (protocol 1, Supporting Information);
Ref (20).
IC50 value = 113 ± 9 μM; NT stands for not tested.
Figure 3.
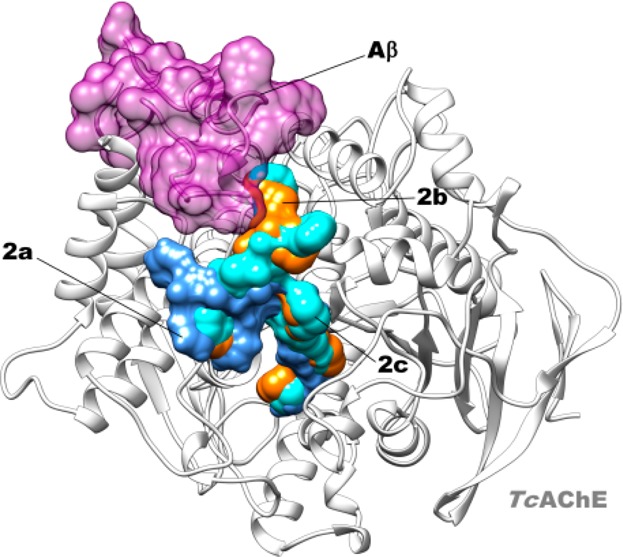
Superposition of the docked pose of Aβ (magenta) in complex with TcAChE (HADDOCK web server version) with IFD poses of 2a (blue), 2b (orange), and 2c (cyan) in complex with TcAChE.
Scheme 1. Synthesis of Compound 2a.

Reagents and conditions: (a) K2CO3, (±)-BINAP, Pd(OAc)2, and 1,8-diaminooctane (for 4), 1,4-dioxane, reflux, 12 h, 62–65%; (b) H2SO4/HNO3, from 0 °C to rt, 30′, 50%; (c) SnCl2·2H2O, EtOH, rt, 12 h, 60%; (d) N-Boc-Gly-OH, ethylchloroformate, THF-DCM, from −10 °C to rt, 3 h, 55%; (e) l-Phe-OBn, EDCI, HOBt, TEA, rt, 12 h, 93%; (f) DEA/DCM, rt, 1 h, 99%; (g) Z-Gly-OH, EDCI, HOBt, TEA, rt, 12 h, 65%; (h) HCOOH, rt, 3 h, 99%; (i) (1) MeCOCl, MeOH, rt, 10‘, 99%; (2) 12, EDCI, HOBt, TEA, from 0 °C to rt, 12 h, 42%.
These data were rationalized by means of molecular modeling studies performed on 1c and 2a–c in complex with TcAChE, which allowed us to directly compare results with those obtained from the crystal structure of 1b in complex with TcAChE (PDB: 2CEK)16 (X-ray studies of 2a were performed on TcAChE21), even though the compounds were tested on human proteins.22 We applied the Induced Fit Docking (IFD) protocol.23 The IFD output for 1c is shown in Figure 1SI, Supporting Information and those for 2a–c are shown in Figures 2 and 2SI, Supporting Information. The reported docked solutions, belonging to the most populated clusters, show very high glide XP scores (higher than those obtained for the other clusters; Table 1SI, Supporting Information) thus representing the most reliable binding mode to TcAChE. By comparison of the X-ray complex of 1b and dockings of 2a–c with TcAChE, we observed that while the bis-tacrine moieties of 2a–c spans the gorge as 1b and other bis-tacrine inhibitors, the distal peptide-like moiety binds on the surface of the enzyme, in the vicinity of the PAS with different binding modes (Figure 2 A vs B, C). Furthermore, the terminal tacrine moiety of 2a–c stacks in front of W279 (Figure 2). F330 undergoes a conformational change with the side chain rotated with respect to the native enzyme, in analogy to one alternate conformation of F330 in 2CEK. In contrast to 2CEK, where the W279 is rotated of 90°, our computational approach revealed that W279 nearly overlaps the native conformation when 2a–c are bound (Figure 3SI, Supporting Information). The replacement of a methylene with an NMe in the linker between the tacrine moieties (2b,c) allows the formation of an additional polar interaction with the key midgorge residue D72 not observed in the complex with 2a (Figure 2, top panels). Besides hydrophobic contacts with F330, Y70, Y121, and Y334, there is one H-bond between the protonated N of the CAS-interacting tacrine of 2a–c and H440 (Figures 2 and 2SI, Supporting Information). Our computational analysis also indicated that the interactions with D72 may be responsible for the substantial differences observed in the disposition of the aromatic moieties of the peptide-like substructure of our compounds at the surface level. Particularly, for 2a all the aromatic moieties project toward the same direction with the benzyl ester engaged in hydrophobic interactions with P337 and L358. Contrarily, for compounds 2b,c, the same interactions are maintained by the benzylcarbamate moiety while the phenylalanine group and the benzyl ester moiety point toward an opposite direction (Figure 2, bottom panels). It is worthy of note that these moieties, although solvent exposed, lay in close proximity to P283 and F284 (Figure 2 bottom panels B–C), which have already been identified as relevant for binding Aβ.8 This evidence is in line with the higher experimentally determined potency of 2b,c in inhibiting hAChE induced Aβ aggregation with respect to 2a. As shown in Figure 3, the peptidic aromatic groups of 2b,c may physically hamper Aβ binding to the surface of the enzyme. Moreover, the peptide-like structure of 2b,c is maintained anchored to the surroundings of the PAS by H-bonds established between a carbonyl and two NH groups with S286 and D285, respectively, for 2b, while for 2c only H-bonds with S286 backbone and side chain were established by the Gly carbonyl group of the ligand (Figures 2 and 2SI, Supporting Information). For bisfunctional 1a and 1c, we confirmed that rotation of W279 at PAS may contribute to the inhibition potency of hAChE-induced Aβ aggregation since W279 is one of the residues critical for Aβ binding. In the case of peptide-based inhibitors 2a–c, it becomes relevant the positioning of the aromatic moieties protruding outside the AChE gorge (e.g., 2c, IC50 = 113 μM) since W279 remains in the apoform position (Figure 3SI, Supporting Information).
Molecular modeling (Figures 2 and 3) and biological studies (Table 1) confirmed this hypothesis, being that 2b,c is more potent than 2a against hAChE-induced Aβ1–40 aggregation, while maintaining nM potency for hChEs inhibition. Furthermore, 2a,c were tested for inhibition of Aβ1–42 spontaneous aggregation. Both compounds exhibited good inhibition properties (Table 1). These encouraging data prompted us to further explore the possible mechanism involved in these antifibrillogenic effects. We investigated the ability of 2a to influence the Aβ1–42 oligomerization process. A capillary electrophoresis (CE) approach, associated with transmission electron microscopy (TEM) analysis,24,25 demonstrated that 2a induces a concentration-dependent interference with the oligomerization kinetics, preventing toxic oligomers and fibril formation. We confirmed that 2a can perturb the formation kinetics of oligomeric intermediate species that were hypothesized to be the real effectors of the neurological damage. Figure 4A shows the control electropherograms after Aβ1–42 dissolution (see Supporting Information and Figure 4SI for details), where toxic oligomers increase over time (peak B area % 77.2 ± 0.9 (t0, n = 3) and 91.4 ± 1.6, (6 h, n = 3) at the expenses of peak A, which is completely depleted on the fourth day from solubilization). Nonbranching Aβ fibrils were present in the precipitated sample (Figure 4D). With 200 μM 2a, a stabilization of oligomer growth occurs (peak B area % 82.5 ± 1.1 (t0, n = 3) and 87.2 ± 2.4 (6 h, n = 3)), and on the second day, the oligomeric species were not detected (Figure 4B). Disaggregation of the toxic oligomers and precipitation are accelerated, if compared to Aβ1–42 alone, and amorphous aggregates are formed in place of fibrils (Figure 4E). Figure 4C shows a more potent and concentration-dependent activity (2a, 500 μM). Freshly solubilized Aβ1–42 does not form toxic, high molecular weight aggregates (peak B), while smaller oligomers (peak A) are detectable and stable only within 24 h (not shown). The TEM analysis (Figure 4F) shows amorphous aggregates. The addition of 50 μM 2a revealed amorphous aggregates at the TEM (n = 2, not shown). Thus, it seems that at higher 2a concentrations (Aβ/2a ratio 1:2 and 1:5) a clear, concentration- and time-dependent depletion of higher molecular weight oligomers occurs (Figure 4B–C), which also produces fibrillogenesis inhibition (Figure 4E,F). Further, 2a (200 and 500 μM) is able to disaggregate preformed fibrils, as from TEM analysis (not shown).
Figure 4.

CE and TEM results: (A) control Aβ1–42 (100 μM); (B) Aβ1–42 (100 μM) incubated with 2a (200 μM); (C) Aβ1–42 (100 μM) incubated with 2a (500 μM); (D–F) correspondent TEM images (n = 3), scale bar = 100 nm.
In conclusion, we describe the synthesis and biological characterization of novel multifunctional tools for the development of innovative DMAADs. Compound 2a was able to interfere with Aβ self-oligomerization, while 2c was found as a good inhibitor of hAChE-induced Aβ aggregation. Biological data demonstrate that binding the surface surrounding the PAS is not correlated to a high potency of inhibition of hAChE-induced Aβ aggregation (being 2a a moderate inhibitor). On the contrary, introduction of a midgorge recognition site in the tether between the two tacrines (2b,c) pushes their peptide portion to occupy the surface where P283 and F284 are critical for Aβ binding. Compounds 2a–c are prototypic of a new class of multifunctional ChEs inhibitors characterized by high potency for enzyme inhibition, for inhibition of hAChE-induced Aβ aggregation, and for inhibition of Aβ self-aggregation and oligomerization.
Acknowledgments
We acknowledge Prof. Marco Racchi for toxicity studies on oligomers. Opinions or assertions contained here are private views of the authors and are not to be construed as official or as reflecting true views of the Department of the Army or the Department of Defence.
Glossary
Abbreviations
- Aβ
Amyloid-beta peptides
- AD
Alzheimer’s disease
- ChE
cholinesterase
- AChE
acetylcholinesterase
- DMAAD
disease-modifying anti-Alzheimer’s drug
- BuChE
butyrylcholinesterase
- PAS
peripheral anionic site
- CAS
catalytic site
- TcAChE
Torpedo californica AChE
- hAChE
human AChE
- hBuChE
human BuChE
- (±)-BINAP
(±)-2,2′-bis(diphenylphosphino)-1,1′-binaphthalene
- THF
tetrahydrofuran
- DCM
dichloromethane
- EDCI
1-ethyl-3-(3-dimethylaminoprpyl)carbodiimide
- TEA
triethylamine
- DEA
diethylamine
- CE
capillary electrophoresis
- TEM
transmission electron microscopy
Supporting Information Available
Further CE notes, supplementary figures and table, details for synthesis, analytical data, and biological studies. This material is available free of charge via the Internet at http://pubs.acs.org.
We acknowledge NatSynDrugs, the Italian Ministry of University and Research (Project PRIN 2009Z8YTYC, and 2010M2JARJ_008), Prog. Reg. Lombardia SAL-45 ID17261, and Unirimini S.p.A. for financial support.
The authors declare no competing financial interest.
Supplementary Material
References
- Lauren J.; Gimbel D. A.; Nygaard H. B.; Gilbert J. W.; Strittmatter S. M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli A.; Bolognesi M. L.; Capsoni S.; Andrisano V.; Bartolini M.; Margotti E.; Cattaneo A.; Recanatini M.; Melchiorre C. A small molecule targeting the multifactorial nature of Alzheimer’s disease. Angew. Chem., Int. Ed. 2007, 46, 3689–3692. [DOI] [PubMed] [Google Scholar]
- Sadowski M.; Wisniewski T. Disease modifying approaches for Alzheimer’s pathology. Curr. Pharm. Des. 2007, 13, 1943–1954. [DOI] [PubMed] [Google Scholar]
- McShane R.; Areosa Sastre A.; Minakaran N. Memantine for dementia. Cochrane Database Syst. Rev. 2006, CD003154. [DOI] [PubMed] [Google Scholar]
- Cummings J. L. Treatment of Alzheimer’s disease: current and future therapeutic approaches. Rev. Neurol. Dis. 2004, 1, 60–69. [PubMed] [Google Scholar]
- Greenblatt H. M.; Dvir H.; Silman I.; Sussman J. L. Acetylcholinesterase: a multifaceted target for structure-based drug design of anticholinesterase agents for the treatment of Alzheimer’s disease. J. Mol. Neurosci. 2003, 20, 369–383. [DOI] [PubMed] [Google Scholar]
- Alvarez A.; Opazo C.; Alarcon R.; Garrido J.; Inestrosa N. C. Acetylcholinesterase promotes the aggregation of amyloid-beta-peptide fragments by forming a complex with the growing fibrils. J. Mol. Biol. 1997, 272, 348–361. [DOI] [PubMed] [Google Scholar]
- De Ferrari G. V.; Canales M. A.; Shin I.; Weiner L. M.; Silman I.; Inestrosa N. C. A structural motif of acetylcholinesterase that promotes amyloid beta-peptide fibril formation. Biochemistry 2001, 40, 10447–10457. [DOI] [PubMed] [Google Scholar]
- Soreq H.; Seidman S. Acetylcholinesterase: new roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [DOI] [PubMed] [Google Scholar]
- Bartolini M.; Bertucci C.; Cavrini V.; Andrisano V. beta-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [DOI] [PubMed] [Google Scholar]
- Reyes A. E.; Chacon M. A.; Dinamarca M. C.; Cerpa W.; Morgan C.; Inestrosa N. C. Acetylcholinesterase-Abeta complexes are more toxic than Abeta fibrils in rat hippocampus: effect on rat beta-amyloid aggregation, laminin expression, reactive astrocytosis, and neuronal cell loss. Am. J. Pathol. 2004, 164, 2163–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvesh S.; Cash M. K.; Reid G. A.; Martin E.; Mitnitski A.; Geula C. Butyrylcholinesterase is associated with beta-amyloid plaques in the transgenic APPSWE/PSEN1dE9 mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2012, 71, 2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I.; Karran E.; De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. The amyloid beta peptide: a chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev. 2012, 112, 5147–5192. [DOI] [PubMed] [Google Scholar]
- Butini S.; Campiani G.; Borriello M.; Gemma S.; Panico A.; Persico M.; Catalanotti B.; Ros S.; Brindisi M.; Agnusdei M.; Fiorini I.; Nacci V.; Novellino E.; Belinskaya T.; Saxena A.; Fattorusso C. Exploiting protein fluctuations at the active-site gorge of human cholinesterases: further optimization of the design strategy to develop extremely potent inhibitors. J. Med. Chem. 2008, 51, 3154–3170. [DOI] [PubMed] [Google Scholar]
- Colletier J. P.; Sanson B.; Nachon F.; Gabellieri E.; Fattorusso C.; Campiani G.; Weik M. Conformational flexibility in the peripheral site of Torpedo californica acetylcholinesterase revealed by the complex structure with a bifunctional inhibitor. J. Am. Chem. Soc. 2006, 128, 4526–4527. [DOI] [PubMed] [Google Scholar]
- Pang Y. P.; Quiram P.; Jelacic T.; Hong F.; Brimijoin S. Highly potent, selective, and low cost bis-tetrahydroaminacrine inhibitors of acetylcholinesterase. Steps toward novel drugs for treating Alzheimer’s disease. J. Biol. Chem. 1996, 271, 23646–23649. [DOI] [PubMed] [Google Scholar]
- Camps P.; Formosa X.; Galdeano C.; Gomez T.; Munoz-Torrero D.; Ramirez L.; Viayna E.; Gomez E.; Isambert N.; Lavilla R.; Badia A.; Clos M. V.; Bartolini M.; Mancini F.; Andrisano V.; Bidon-Chanal A.; Huertas O.; Dafni T.; Luque F. J. Tacrine-based dual binding site acetylcholinesterase inhibitors as potential disease-modifying anti-Alzheimer drug candidates. Chem. Biol. Interact. 2010, 187, 411–415. [DOI] [PubMed] [Google Scholar]
- Minarini A.; Milelli A.; Tumiatti V.; Rosini M.; Simoni E.; Bolognesi M. L.; Andrisano V.; Bartolini M.; Motori E.; Angeloni C.; Hrelia S. Cystamine-tacrine dimer: a new multi-target-directed ligand as potential therapeutic agent for Alzheimer’s disease treatment. Neuropharmacology 2012, 62, 997–1003. [DOI] [PubMed] [Google Scholar]
- Bolognesi M. L.; Cavalli A.; Valgimigli L.; Bartolini M.; Rosini M.; Andrisano V.; Recanatini M.; Melchiorre C. Multi-target-directed drug design strategy: from a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against Alzheimer’s disease. J. Med. Chem. 2007, 50, 6446–6449. [DOI] [PubMed] [Google Scholar]
- Sanson B.Dynamique structurale de l’acétylcholinestérase étudiée par cristallographie aux rayons X et par une méthode spectroscopique complémentaire. Ph.D. Thesis, Université Joseph Fourie, Grenoble I, 2009; see http://www.ibs.fr/science/production-scientifique/theses-soutenues-a-l-ibs/?lang=fr. The refined X-ray structure will be reported elsewhere. [Google Scholar]
- Molecular modelling studies on hChEs will be reported elsewhere.
- Schrödinger Suite 2011 Induced Fit Docking; Glide version 5.7; Prime version 3.0; Schrödinger, LLC: New York, 2011.
- Sabella S.; Quaglia M.; Lanni C.; Racchi M.; Govoni S.; Caccialanza G.; Calligaro A.; Bellotti V.; De Lorenzi E. Capillary electrophoresis studies on the aggregation process of beta-amyloid 1–42 and 1–40 peptides. Electrophoresis 2004, 25, 3186–3194. [DOI] [PubMed] [Google Scholar]
- Colombo R.; Carotti A.; Catto M.; Racchi M.; Lanni C.; Verga L.; Caccialanza G.; De Lorenzi E. CE can identify small molecules that selectively target soluble oligomers of amyloid beta protein and display antifibrillogenic activity. Electrophoresis 2009, 30, 1418–1429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.