

Abstract
Human mitogen-activated protein kinases (MAPK)-interacting kinases 1 and 2 (Mnk1/2) are promising anticancer targets. Mnks possess special insertions and a DFD-motif that are distinct from other kinases. Crystallographic studies of Mnk1/2 have revealed that the DFD-motif adopts the DFG/D-out conformation in which residue F227 flips into the ATP binding pocket. This is rarely observed in other kinases. Although the DFG-out conformation has attracted great interest for designing selective inhibitors, structural requirements for binding and the mechanism governing the DFG-out conformation remain unclear. This work presents for the first time the applicability of 3D models of Mnk2 protein in studying conformational changes by utilizing homology modeling and molecular dynamics simulations. The study reveals that the interactions between residue K234 of insertion I1 and D226 of the DFD motif play a key role in inducing and stabilizing the DFD-out conformation. The structural features will aid in the rational design of Mnk2 inhibitors.
Keywords: Mnk, molecular dynamics simulation, DFD motif, insertion I1
Human mitogen-activated protein kinases (MAPK)-interacting kinases 1 and 2 (Mnk1 and Mnk2) modulate the function of eukaryotic initiation factor 4E (eIF4E) through phosphorylation. eIF4E plays a key role in the PI3K/Akt/mTOR- and Ras/Raf/MEK/ERK-mediated tumorigenesis. Hence, Mnk1/2-mediated eIF4E phosphorylation is crucial for oncogenic activity.1,2 Importantly, Mnk seems to be dispensable for normal development.3 Targeting Mnk may, therefore, provide a therapeutic avenue for nontoxic anticancer drug development.4
Understanding the structure and structural plasticity of Mnks is essential to design new inhibitors, especially in targeting the two different conformational states of Mnk. Inspection of the crystal structures of nonphosphorylated Mnk1/2 kinase domains (Mnk1/2-KD)5,6 reveal the bilobed (i.e., N- and C-terminal lobes) makeup of arrangement that is folded by secondary structural elements, connecting through a set of hinge residues. The ATP binding pocket is a hydrophobic cleft between the two lobes.5,6 The N-terminal lobe is divided into 4–5 antiparallel β-sheets (β1−β5) and a regulatory helix αC. This region harbors the highly flexible glycine-rich loop that is critical for ATP adenine ring binding. Additionally, conserved Lys and Glu residues in this lobe are essential for the stabilization of ATP substrates. The C-terminal lobe, in contrast, consists of predominantly hydrophobic α-helical bundles. It harbors the three key elements; catalytic loop, magnesium-binding loop, and activation loop (A-loop), which are required for phosphate transfer, magnesium ion coordination, and substrate binding, respectively.5,6
Mnks possess two distinct features: (1) a noncanonical DFD-motif (Asp191–Phe192–Asp193 in human Mnk1 and Asp226–Phe227–Asp228 in human Mnk2) that lie within the magnesium-binding loop, replacing the DFG (Asp–Phe–Gly) motif typically found in other kinases; and (2) the catalytic domain contains three Mnk specific inserts (i.e., insertions I1, I2, and I3) not observed in other kinases.6 The DFD fingerprint is unique within the human kinome. Furthermore, this DFD motif adopts an unusual DFD-out conformation, in which the Phe residue flips into the ATP binding pocket, prevents the accessibility of ATP,6 and exposes an additional hydrophobic/allosteric site. Among more than 50 protein kinases for which structures are available, only 10% have been observed with the DFD-out conformation. More rarely, Mnk adopts the DFD-out conformation in the absence of ligands (PDB 2AC3). To date, there is no crystal structure with Mnk1/2 adopting the DFD-in conformation. Insertion I1 is also of particular interest as this short residue sequence (I233K234LNGDCS240) is located within the A-loop, at only 4 residues away from the unique DFD-motif. However, the role of this specific insertion I1 has remained elusive.
Studies have shown that a productive inactive-to-active transition typically involves the orientation changes of two key elements: (1) the A-loop in the C-terminal lobe and (2) the helix αC in the N-terminal lobe.7 Flipping of the DFD motif between DFD-in (open/active) and DFD-out (closed/inactive) conformations has been suggested to facilitate the binding and release of ATP or inhibitor.8 However, the mechanism in which Mnk adopts the DFD-out conformation in its apo form remains a question of considerable interest. Advances in understanding the mechanism underlying the DFD flip would enhance the understanding of kinase activation and open new directions for designing a selective inhibitor. As it is very challenging and cost-consuming to monitor the dynamics of the DFD motif experimentally, computational approaches using molecular dynamics (MD) simulations can provide useful insights.
MD simulation has been extensively used for the studies of conformational rearrangements and the time-dependent behavior of biomolecular systems. However, a major drawback of using traditional MD simulations to study large-scale conformational changes in biomolecules is that it suffers from inadequate sampling due to difficulties in surmounting multiple energy barriers in affordable computational time. The time scales for protein side chains and backbone movements are typically ∼10–9 and ∼10–5 s, respectively, which entails a microsecond time scale for the process of interconversion between an inactive and active kinase.9,10 For example, the DFG-flip of the Abl protein was observed only once in 2 μs in a standard MD simulation, and it occurred only after mutation.9 By performing high-temperature MD simulation,11−14 applying temperature-accelerated molecular dynamics (TAMD)15 or using an addictive biasing potential to flatten the free energy profile, the problem of free energy barriers can be potentially overcome, while avoiding the long waiting times inherent in traditional MD.7
In the present study, high-temperature MD simulation was applied to Mnk2 and Mnk2 mutant models to investigate the underlying mechanism why the DFD-out conformation is preferable and to identify the conformational changes between the DFD-out and -in variants. Since none of the available crystal structures present the A-loop (residues 232–250), homology model was built to restore the missing A-loop and the so-called insertion I3, ranging from residues 304–310 (Figure 1).
Figure 1.
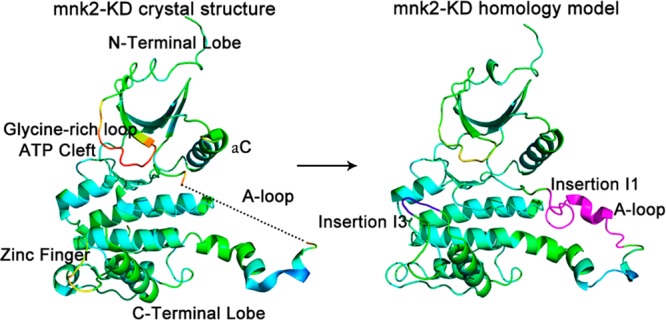
X-ray crystal structure of the Mnk2-KD (PDB entry 2AC3) and its homology model, in which the missing activation loop (purple, residues 232–250) and insertion I3 (blue, residues 304–310) were built. The homology model is generated using the crystal structure as a template, followed by multiple-threading alignments with I-TASSER (Table S1, Supporting Information). On the basis of the homology model, other Mnk2 mutant models were generated by Modeler.
In order to depict the differences in dynamics of the DFD flip between the inactive and active conformations, both the closed (i.e., DFD-out) and open (i.e., DFD-in) states of Mnk2 and Mnk2 mutant homology models were built and studied in parallel. First, Mnk2-in and Mnk2-out models were built to restore the missing regions as mentioned above. Second, since previous crystallographic studies have revealed that a single mutation of Asp228 (D228) to a glycine residue allows both conformations to be adopted,5 two models, Mnk2DFG-in and Mnk2DFG-out (where DFD228 has been replaced by DFG228) were built to investigate how residue D228 contributes to the DFG/D-out conformation. Third, as mentioned in the introduction, Mnk-specific insertion I1 can potentially play a role in the A-loop conformation as well as the DFG/D-out conformation. Hence, Mnk2mutant-in and Mnk2mutant-out models, in which the specific insertion I1 has been removed, were also developed. Lastly, it is also of interest to study the combined effect of the above-mentioned mutations, therefore both Mutant-in and Mutant-out models, in which the DFD-motif was replaced by DFG and the insertion I1 was removed, were also constructed in this study. A total of eight models used in the study are summarized in Table S2, Supporting Information.
In this study, the crystal structure of Mnk2 protein (PDB 2AC5) was adopted as a template to investigate the conformational transitions of the DFD-motif between the DFD-in and DFD-out conformations. The atomic distance between the phenyl ring of the gatekeeper Phe159 and the Phe227 of the DFD-motif (DF159–F227) for both conformations was measured, and as shown in Figure 2A, DF159–F227 is 5.2 Å when the DFD-out conformation of Mnk2 was adopted, whereas the DFD-in conformation has a DF159–F227 of 9.8 Å. Following this, the time-evolution of DF159–F227 of the DFD-motif with reference to the homology models (Figure 1) built for the simulation studies was investigated, as shown in Figure 2B-2I, in order to determine the respective conformational changes of the DFG/D motif. This step can be easily achieved by executing a script containing the relevant atomic information in the PTRAJ module of the Amber12 package.16
Figure 2.
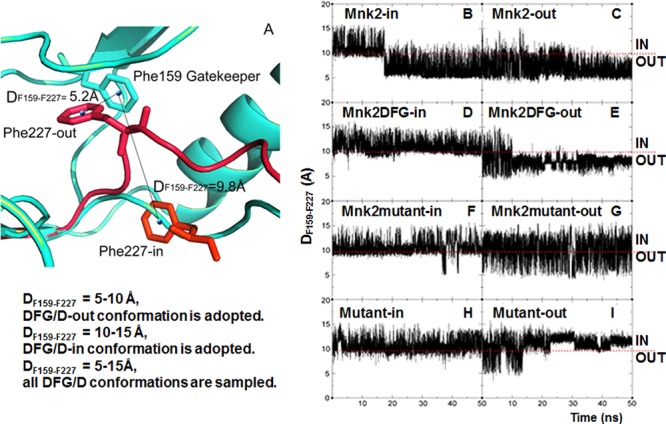
(A) Interatomic distance between the phenyl ring of the gatekeeper F159 and F227 of the DFD-motif (DF159–F227) for both DFD-in/-out conformations using the Mnk2 crystal structure (PDB 2AC5) as the template. (B–I) Time-evolution of the DF159–F227 of the DFD-motif with reference to the homology models built for the simulation studies. The DF159–F227 versus simulation time (ns) plots were analyzed using the PTRAJ module in the AMBER suite, while trajectories were visualized and validated using VMD software.17
Visual inspection of the resulting trajectories in conjunction with distance monitoring suggests that when DF159–F227 = 5–10 Å, the DFG/D-out conformation was adopted; when DF159–F227 = 10–15 Å, the DFG/D-in conformation was adopted; and when DF159–F227= 5–15 Å, all the DFG/D conformations were sampled. As the F159 was stable (Figure S1, Supporting Information), the schemes were found consistently in all simulations and are used as the basis for further discussion. It is worth noting that the Mnk2-in model was initially simulated at a temperature of 300 K, but no appreciable conformational change of the DFD-motif was observed in a 200 ns simulation (Figure S2, Supporting Information), indicating that the time scale for DFD flip is much longer than a few hundred nanoseconds. This is not surprising since large conformational rearrangements, such as the opening/closing of the kinase domain receptor (KDR) domain, are too slow to occur.7
All the models were then subjected to a high temperature simulation (1000 K). The results are shown in Figure 2. Moreover, all the simulated models were repeated twice for verification and the second set of results are shown in Figure S3, Supporting Information. A sudden drop of DF159–F227 from 10–15 Å to 5–10 Å at 17 ns (at 11 ns in the repeated simulation, Figure S3A, Supporting Information) was observed for the Mnk2-in model (Figure 2B), indicating a DFD flip from DFD-in to -out conformation. The DFD-out conformation was observed to remain stable for the rest of the simulation. For the Mnk2 model starting from the DFD-out conformation, a small jump of DF159–F227 at approximate 14 ns was observed (Figure 2C). Visual inspection of the trajectory at this time-point reveals a flip from the DFD-out to -in conformation; however, the DFD-in conformation lasted for less than 100 ps of the simulation time, and then reverted back to the DFD-out conformation. During the last 36 ns of simulation time, the DFD-out conformation was maintained and stabilized as indicated by the DF159–F227 of 5–10 Å. For the repeated simulation of this model, the DFD-out conformation was maintained after 5 ns. These results suggest that Mnk2 prefers to adopt an inactive DFD-out conformation, being consistent with the observation of the DFD-out conformation present in known crystal structures.5 The results are reproducible as suggested by repeated simulations (Figure S3A,B, Supporting Information).
As shown in Figure 2D, the starting DFG-in conformation in the Mnk2DFG mutant model remained stable over the course of the simulation, as indicated by the DF159–F227 of 10–15 Å. However, Figure 2E highlights that the DF159–F227 oscillates between 5 and 15 Å in the first 10 ns simulation of the Mnk2DFG-out model, indicating that all DFG conformations were sampled. However, the DFG-out conformation was stabilized after 10 ns of equilibration, in which the DF159–F227 value converged to 5–10 Å. The repeated study (Figure S3D, Supporting Information) displayed a similar trend as for that shown in Figure 2E. Together, these results are in agreement with the crystallographic study suggesting that both DFG-in and DFG-out conformations were adopted upon the mutation of Asp228 to Gly228 in Mnk2.5 For the Mnk2 mutant model starting from the DFG-in conformation where insertion I1 has been removed, several decrements of DF159–F227 were observed in the range 35–42 ns, as shown in Figure 2F. Visual inspection of the trajectories revealed that the DFD-motif flips between the DFD-in and -out conformations during the 35–42 ns time period, then adopts the DFD-in conformation for the rest of simulation. In contrast, the Mnk2mutant-out model experiences a larger fluctuation for the DFD-motif with DF159–F227 values of 5–15 Å observed during the entire simulation (Figure 2G), indicating sampling of all DFD conformations. These observations are further supported by similar plots obtained in the repeated work as shown in Figure S3E,F, Supporting Information, suggesting reproducibility of simulations. The results, taken in totality, suggest that the DFD-motif is relatively more flexible compared to other models upon removal of insertion I1.
Interestingly, upon the combined mutations of D228G and the removal of insertion I1, simulations in either closed or open starting conformation show that the DFG-motif prefers to stay in the DFG-in conformation for the entire run, with the exception of the first 15 ns in the Mutant-out model (Figure 2I), which might be explained by the equilibration time required by the model.
To measure the flexibilities of the A-loop during MD simulations, the root mean-square deviation (rmsd) values of the A-loop backbone atoms (N,CA,C) were monitored for all MD trajectories and compared with the corresponding starting structures (Figure S4, Supporting Information). Except for the Mutant-in model, the rmsd values for all models oscillate to ∼10 Å within the first few ns of MD-equilibration and reach a plateau for the rest of the simulation. The rmsd values for the Mutant-in model were found to have lower values oscillating around 5 Å, suggesting the A-loop is highly stable and does not move significantly away from its initial active-like conformation (Figure S4, Supporting Information).
To confirm some general considerations of A-loop conformation, a comparison between starting models and stimulated structures for all models was performed. Each simulated structure, also known as the averaged structure, was developed by averaging 2000 MD simulation snapshots taken from the last 4 ns of the corresponding trajectories. As shown in Figure S5, Supporting Information, the A-loop in the Mutant-in model stayed in an open conformation, while the A-loop in other models escaped from the open conformation free energy basin, reaching an intermediate structure between the open and closed conformations. The shape adopted was found to be very similar to the MD simulation studies of CDK5, Hck, c-Src, and Abl protein kinases.7,9,18,19 It is worth noting that once the A-loop in each model reached the intermediate state, this conformation was maintained over the course of simulations without flipping to a fully open/closed conformation.
To provide a more detailed picture of the local changes occurring during the simulated DFD flip in the Mnk2-in model, snapshots were taken at six different time-points as shown in Figure 3. Phe227 leaves the buried hydrophobic pocket (also referred to as the hydrophobic spine) and flips clockwise to a solvent-exposed pocket by about 180°, thus adopting the DFD-out conformation as observed in the crystal structure. Previous findings suggested that the DFG/D-motif phenylalanine is part of the hydrophobic spine, which stabilizes the DFG/D-in conformation.20 Consistent with this, closer inspection of the Mnk2-in model revealed a cavity around the DFD-motif (as shown in blue mesh in Figure 3) in the DFD-out conformation but not in the DFD-in conformation. The spine created by hydrophobic residues, including β4 leucine, helix αC leucine, DFG-phenylalanine, and HRD-tyrosine,20 is known to perform coordinated motions facilitating the open–closed conformational switch during the catalytic cycle for most kinases.21 Hence, a question arises as to why the DFD-out conformation was favorable for the rest of the simulations.
Figure 3.
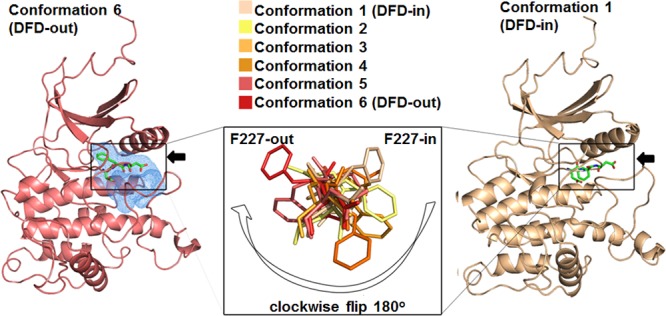
Conformational changes involved in the DFD flip. Snapshots (as shown in the inset) were taken from the trajectory at 17.13, 17.17, 17.21, 17.29, 17.33, and 17.34 ns in the Mnk2-in model. As indicated by the white arrow, starting from conformation 1 (the active DFD-in conformation, right-hand structure), Phe227 leaves the buried hydrophobic pocket and flips clockwise to a solvent-exposed pocket by about 180° (conformation 6, the inactive DFD-out conformation, left-hand structure). The hydrophobic pocket spine is shown in blue mesh. The arrows indicate the side views of the DFD motif.
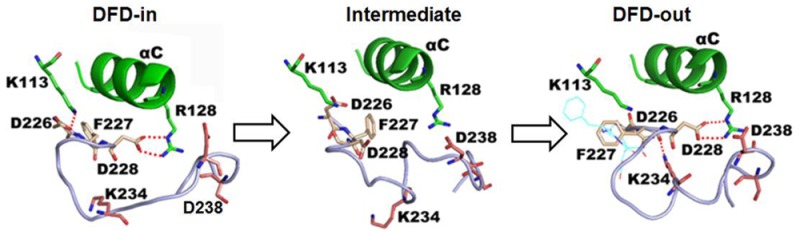
To elucidate the interactions that induce and stabilize the DFD-out conformation, the averaged structures taken from before, during, and after the DFD flip in Mnk2-in were compared, as shown in Figure 4. Before the DFD flip, residues Asp226 and Asp228 of the DFD-motif formed a salt bridge with the β3 strand Lys113 and the αC helix Arg128, respectively (Figure 4A). These salt-bridge interactions were broken during the DFD flip (Figure 4B), but Asp228 re-established the interaction with Arg128 upon completion of the transition (Figure 4C). Interestingly, a new interaction between Asp226 and Lys234 was observed after the DFD flip. Monitoring the interatomic distances of salt-bridge interactions showed that there is a competition between Lys113 and Lys234 for Asp226. Initially, interaction between Asp226 and Lys113 is more stable than Asp226–Lys234. Upon DFD flip, Lys234 is outcompeting Lys113, which signifies the stronger ability of Lys234 to form a salt bridge interaction with Asp226 relative to Lys113. The salt bridge interaction is stabilized over the course of simulation after the flip (Figure 4D,E). These results imply that the salt bridge between Asp226 and Lys234 plays an important role in inducing the DFD flip and contributes to the stabilization of the DFD-out conformation. In addition, superposition of the initial structure and the averaged structure taken from the last 4 ns trajectory (Figure 4C) showed that the conformation of Asp226 was well preserved, while Asp228 was engaged in forming a salt bridge interaction with Arg128; therefore, Asp228 moves away from its initial position and points toward helix αC. Consistently, the model Mnk2-out is maintained in the DFD-out conformation for the entire simulation, and the salt-bridge interaction of Asp226–Lys234 was also observed (Figure S6A,B, Supporting Information). Monitoring the Asp226–Lys113 distance in the Mnk2-out model showed that it fluctuates at the same level as the distance shown in Figure 4D,E after the flip, indicating that such an interaction is weaker than Asp226–Lys234. Together, these results suggest that the DFD-out conformation is preferable in Mnk2 and that Lys234 plays an important role in the stabilization of the DFD-out conformation.
Figure 4.
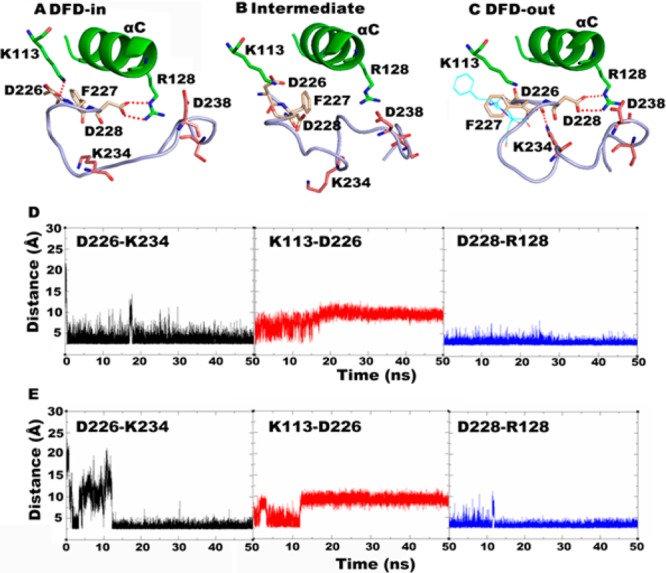
All simulation structures and data shown are taken from the Mnk2-in model. (A) Averaged structure taken from trajectories at 6–10 ns. (B) Snapshot taken from the trajectory at 11.58 ns. (C) Superimposition of initial structure (cyan sticks) and averaged structure taken from the last 4 ns of trajectory time. (D) Interatomic distances for key salt-bridge interactions during the simulation. (E) Interatomic distances for key salt-bridge interactions taken from the repeated simulation. The DFD-motif is shown in yellow sticks; insertion I1 is shown in pink sticks; salt-bridge interactions are shown by red dotted lines.
However, for models Mnk2DFG-in and Mnk2DFG-out, a salt bridge between Asp226 and Lys234 was not observed (Figure S7, Supporting Information). Overlay of structures revealed a slight rotation movement of the DFG-motif in both models. Together, these observations could be explained by the lack of an Asp228 salt bridge interaction, resulting in a rotation movement of the DFG-motif, preventing the formation of the Asp226–Lys234 salt bridge. Interestingly, the direction of DFG rotation in the Mnk2DFG-in model is opposite to the Mnk2DFG-out model (as shown by the red and blue arrows in Figure S7, Supporting Information). For models Mnk2mutant-in and Mnk2mutant-out, the averaged structures shown in Figure S8 displayed a larger shift of the DFD-motif than the models in Figure S7, Supporting Information. Similarly, without insertion I1, no salt bridge could be established between Asp226 and Lys234 (Figure S8, Supporting Information).
For the Mutant-in model, superposition of the structures showed that the DFG-in conformation was well preserved (Figure S9A, Supporting Information), whereas a conversion from the DFG-out to the DFD-in conformation was observed in model Mutant-out (Figure S9B, Supporting Information). A salt bridge between Asp226 and Lys113 was also observed in both models, highlighting the importance of such a salt bridge in maintaining the DFG/D-in conformation. The results once again indicate the important role played by Asp228 of the DFD motif and Lys234 of insertion I1 in stabilizing the DFD-out conformation of Mnk2.
On the basis of the simulation results, an atomistic mechanism of how the DFD-motif and insertion I1 stabilize the DFD-out conformation in Mnk2 protein kinase is proposed. The DFD-out conformation is highly favorable in Mnk2 due to the presence of an internal salt-bridge interaction (Lys234–Asp226) within the activation loop (Figure 4C). Such an interaction plays a key role in controlling the flexibility of the A-loop. Clockwise rotation of the DFD-motif involves the breakage of the Asp226–Lys113 salt bridge interaction, where a new interaction between Asp226 and Lys234 is established upon completion of the DFD flip (Figure 4C). Removing insertion I1 eliminated the Asp226–Lys234 salt bridge; in this circumstance, Asp226 can only form an interaction with Lys113, and hence, DFD flip cannot be induced (Figures S8 and S9, Supporting Information). DFD flip also involves the breakage of the salt bridge between Asp228 and Arg128, but the interaction is restored immediately after the flip is complete. It is hypothesized that the interaction is first broken during the DFD flip, anchoring itself with either Lys113 or Arg128 after the flip and assisting the Asp226–Phe227 flip (Figure 4). Mutation of Asp228 to Gly228 causes Asp226 to take over the role of Asp228, flipping back and forth without fixed direction (Figure S7, Supporting Information) and forming salt bridge interactions with either Lys113 or Arg125, resulting in both active and inactive conformations (Figures S7 and S9, Supporting Information). In particular, the DFD-in conformation is adopted when the Asp226–Lys113 salt bridge forms (Figures S7A, S8A, and S9, Supporting Information). Single mutation of either DFD228G or removed insertion I1 has little impact on the DFD flip in comparison to the combined mutations (Figures S7 and S8, Supporting Information). Double mutations further increased the DFG-motif flexibility. The formation of salt bridge Asp226–Lys113 destabilized the DFG-out conformation to the advantage of the DFG-in conformation.
MD simulation studies in other protein kinases, such as IRK and VEFGR2, suggested that the DFG/D flip is associated with conformational change (opening/closing) of the A-loop and is driven by a balance of concurrent electrostatic and hydrophobic interactions.7,15 Mutation of models in the present study showed folding of the A-loop into a three-turn-fold intermediate conformation, which further facilitating DFG/D-flip, is in agreement with the findings as described. The DFG/D-motifs are highly mobile, and the movement of the A-loop to the DFG-out conformation exposes an additional hydrophobic binding site (referred to as the allosteric site), which is directly adjacent to the ATP binding site. Type II inhibitors such as imatinib and BIRB796 can bind to both the ATP binding site and allosteric site, exhibiting higher selectivity and resulting in better safety profiles. Crystallographic studies have supported the hypothesis of a DFG-out conformation induced by allosteric inhibitors.22 NMR studies of the dynamic equilibrium between the DFG-in and DFG-out conformations of p38α suggested that the DFG-motif in apo p38 is in conformational exchange between DFG-in and -out; however, the DFG-out conformation is sampled less frequently than DFG-in conformation.23 Inhibitors that bind to the DFG-out form freeze the protein in the DFG-out conformation and suppress the conformational exchange. The present study indicates that, in the case of Mnk2, the DFD-out conformation is sampled more frequently than the DFD-in conformation and that the design of DFD-out ligand against Mnk2 offers a promising way forward.
The application of conversional high-temperature MD simulation is highly effective for the investigation of biologically relevant conformations in proteins. It is capable of providing new structural insights not found in crystal structures. Such MD simulation types do not require the artificial weakening of interactions between a set of residues that may induce uncontrolled conformational rearrangements. However, high-temperature MD simulation suffers from partial loss of hydrophobic interactions stabilized by water.
Overall, our findings are in agreement with previous crystallographic studies and provide new insights into the DFD conformation of Mnk2. Further studies of mutagenesis of the key residues including Lys234 will be the subject of future communications.
Supporting Information Available
Material detailing methods and modeling-derived information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Buxade M.; Parra-Palau J. L.; Proud C. G. The Mnks: MAP kinase-interacting kinases (MAP kinase signal-integrating kinases). Front. Biosci. 2008, 13, 5359–5373. [DOI] [PubMed] [Google Scholar]
- Waskiewicz A. J.; Flynn A.; Proud C. G.; Cooper J. A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997, 16, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N. Mnk earmarks eIF4E for cancer therapy. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 13975–13976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J.; Lam F.; Proud C.; Wang S. Targeting Mnks for cancer therapy. Oncotarget 2012, 3, 118–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauch R.; Jakel S.; Netter C.; Schreiter K.; Aicher B.; Jackle H.; Wahl M. C. Crystal structures of the Mnk2 kinase domain reveal an inhibitory conformation and a zinc binding site. Structure 2005, 13, 1559–1568. [DOI] [PubMed] [Google Scholar]
- Jauch R.; Cho M. K.; Jakel S.; Netter C.; Schreiter K.; Aicher B.; Zweckstetter M.; Jackle H.; Wahl M. C. Mitogen-activated protein kinases interacting kinases are autoinhibited by a reprogrammed activation segment. EMBO J. 2006, 25, 4020–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chioccioli M.; Marsili S.; Bonaccini C.; Procacci P.; Gratteri P. Insights into the conformational switching mechanism of the human vascular endothelial growth factor receptor type 2 kinase domain. J. Chem. Inf. Model. 2012, 52, 483–491. [DOI] [PubMed] [Google Scholar]
- Kannan N.; Neuwald A. F. Did protein kinase regulatory mechanisms evolve through elaboration of a simple structural component?. J. Mol. Biol. 2005, 351, 956–972. [DOI] [PubMed] [Google Scholar]
- Shan Y.; Seeliger M. A.; Eastwood M. P.; Frank F.; Xu H.; Jensen M. O.; Dror R. O.; Kuriyan J.; Shaw D. E. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman B. F.; Lipson D.; Wemmer D. E.; Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science 2001, 291, 2429–2433. [DOI] [PubMed] [Google Scholar]
- Frembgen-Kesner T.; Elcock A. H. Computational sampling of a cryptic drug binding site in a protein receptor: explicit solvent molecular dynamics and inhibitor docking to p38 MAP kinase. J. Mol. Biol. 2006, 359, 202–214. [DOI] [PubMed] [Google Scholar]
- Bruccoleri R. E.; Karplus M. Conformational sampling using high-temperature molecular dynamics. Biopolymers 1990, 29, 1847–1862. [DOI] [PubMed] [Google Scholar]
- Pande V. S.; Rokhsar D. S. Molecular dynamics simulations of unfolding and refolding of a beta-hairpin fragment of protein G. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9062–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimi N. L.; Ho B.; Agard D. A. Unfolding simulations reveal the mechanism of extreme unfolding cooperativity in the kinetically stable alpha-lytic protease. PLoS Comput. Biol. 2010, 6, e1000689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashisth H.; Maragliano L.; Abrams C. F. ″DFG-flip″ in the insulin receptor kinase is facilitated by a helical intermediate state of the activation loop. Biophys. J. 2012, 102, 1979–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T.E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Hayik S.; Roitberg A.; Seabra G.; Swails J.; Goetz A. W.; Kolossváry I.; Wong K. F.; Paesani F.; Vanicek J.; Wolf R. M.; Liu J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Wang J.; Hsieh M.-J.; Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Salomon-Ferrer R.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. A.. AMBER 12; University of California: San Francisco, CA, 2012.
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Berteotti A.; Cavalli A.; Branduardi D.; Gervasio F. L.; Recanatini M.; Parrinello M. Protein conformational transitions: the closure mechanism of a kinase explored by atomistic simulations. J. Am. Chem. Soc. 2009, 131, 244–250. [DOI] [PubMed] [Google Scholar]
- Gan W.; Yang S.; Roux B. Atomistic view of the conformational activation of Src kinase using the string method with swarms-of-trajectories. Biophys. J. 2009, 97, L8–L10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jura N.; Zhang X.; Endres N. F.; Seeliger M. A.; Schindler T.; Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 2011, 42, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev A. P.; Haste N. M.; Taylor S. S.; Eyck L. F. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 17783–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [DOI] [PubMed] [Google Scholar]
- Vogtherr M.; Saxena K.; Hoelder S.; Grimme S.; Betz M.; Schieborr U.; Pescatore B.; Robin M.; Delarbre L.; Langer T.; Wendt K. U.; Schwalbe H. NMR characterization of kinase p38 dynamics in free and ligand-bound forms. Angew. Chem., Int. Ed. 2006, 45, 993–997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.