

Abstract
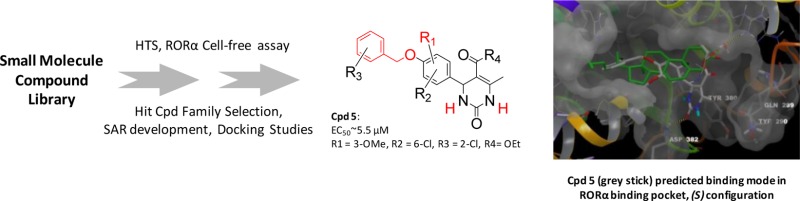
A high throughput screen was developed to identify novel, nonsteroidal RORα agonists. Among the validated hit compounds, the 4-(4-(benzyloxy)phenyl)-5-carbonyl-2-oxo-1,2,3,4-tetrahydropyrimidine scaffold was the most prominent. Among the numerous analogues tested, compounds 8 and 9 showed the highest activity. Key structure–activity relationships (SAR) were established, where benzyl and urea moieties were both identified as very important elements to maintain the activity. Most notably, the SAR were consistent with the binding mode of the compound 8 (S-isomer) in the RORα docking model that was developed in this program. As predicted by the model, the urea moiety is engaged in the formation of key hydrogen bonds with the backbone of Tyr380 and Asp382. The benzyl group is located in a wide hydrophobic pocket. The structural relationships reported in this letter will help in further optimization of this compound series and will provide novel synthetic probes helpful for elucidation of complex RORα physiopathology.
Keywords: RORα, NR1F1, agonist, docking, SAR, Biginelli
The retinoic acid-related orphan receptor alpha (NR1F1, RORα) is a ligand-activated transcription factor and a member of the nuclear hormone receptor superfamily. The characterization of RORα-deficient mice (staggerer mice)1−3 that carry the natural, nonsense mutation in the NR1F1 gene has provided a great insight into the critical functions of RORα in the regulation of a variety of physiological and developmental processes, related to bone health, energy homeostasis, immune function, inflammatory responses, brain development processes, and neuroprotection. RORα activity modulates the expression of several components of the circadian clock system. This receptor is also suspected to play a role in integrating the central pacemaker signal and the rhythmic expression pattern of downstream (metabolic) genes. For a thorough description of RORα-related physiology, readers are referred to the recent excellent reviews.4−7
Kallen et al. described the first crystal structure of the ligand binding domain (LBD) of RORα. Surprisingly, authors identified a molecule of cholesterol that was present in the ligand binding pocket of the RORα protein that was purified from insect cells.8 They concluded that cholesterol (or its derivative) binding in RORα LBD stabilized the receptor in the agonistic conformation. Later, they further demonstrated that cholesterol sulfate also binds within the pocket, although with better affinity.9 Recently, 7-α-hydroxycholesterol, 7-β-hydroxycholesterol, and 24-S-hydroxycholesterol were identified as RORα inverse agonists.10,11 These novel ligands bound to the target protein with nanomolar affinities, down-regulated RORα activity in reporter assays, and down-regulated target gene expression in vitro. The interpretation of dynamic hydrogen/deuterium exchange experiments (HDX) on the ligand–receptor complex led to the model in which the unliganded RORα-LBD (protein produced in bacteria that do not contain cholesterol) is in an active conformation and constitutively interacts with the coactivator peptide. The binding of an inverse agonist ligand, such as 7-α-hydroxycholesterol leads to a decrease in the affinity for the coactivator binding and results in a decreased transactivation activity and reduced transcriptional output.11
Finally, two synthetic RORα ligands have been recently published.12,13 These small molecules, SR1078 (dual RORα/γ agonist) and SR3335 (RORα selective inverse agonist), modulated RORα activity in reporter assays and target gene expression in cell culture models.
In this letter, we report on the discovery of new nonsteroidal RORα ligands by high throughput screening of a small molecule compound library and on the preliminary optimization of one hit series (Hit Scaffold 1, Scheme 1). In particular, the structure–activity relationships (SAR) that corroborate the results of the docking studies with the most active compounds (in-house docking model) will be highlighted and further optimization opportunities will be pointed out.
Scheme 1. General Synthetic Pathway to 4-(4-(Benzyloxy)phenyl)-5-carbonyl-2-oxo-1,2,3,4-tetrahydro-pyrimidine to Hit Scaffold 1.

The screen was accomplished in a cell-free format with the use of the pull-down assay that measured the recruitment of the TIF2-BAP reporter hybrid protein on the immobilized GST-RORα protein. TIF2 (NCOA2) was chosen for this project since it was previously described to serve as a natural RORα coactivator with a distinct physiological function.14 The recruitment assay was validated with a known RORα ligand, 7-dehydro-cholesterol (7DC, cf. Supporting Information Supplementary Figure 1).8
The pull-down assay was robust, reproducible, and easily automated. The primary screen was accomplished with a hit rate of 0.16% and with a satisfactory z′ factor of 0.55. In addition to steroid compounds, various small molecules were identified as RORα ligands. A specific activity of all these compounds was confirmed in dose response studies with a TIF2-BAP protein but no interaction was found with a TIF2(mut)-BAP protein, where all the three LXXLL motifs were invalidated by side directed mutagenesis. In addition, none of the hits were active in counter-screen assays, which scored for nonspecific interactions, such as direct GST binding or BAP (reporter protein) activation (not shown).
Following the assignment (structure related) of each confirmed hit to a specific chemical series, singletons and hit series (comprising several hits) were identified. A hit series evaluation process began, considering drug-like properties, synthetic accessibility, SAR, etc, and analogues or compounds complementary to the chemical space around these series, either purchased, synthesized, or identified within our library, were tested in dose–response studies.
In the case of Scaffold 1 (Scheme 1), some 300 related analogues, including several additional active compounds, were tested.
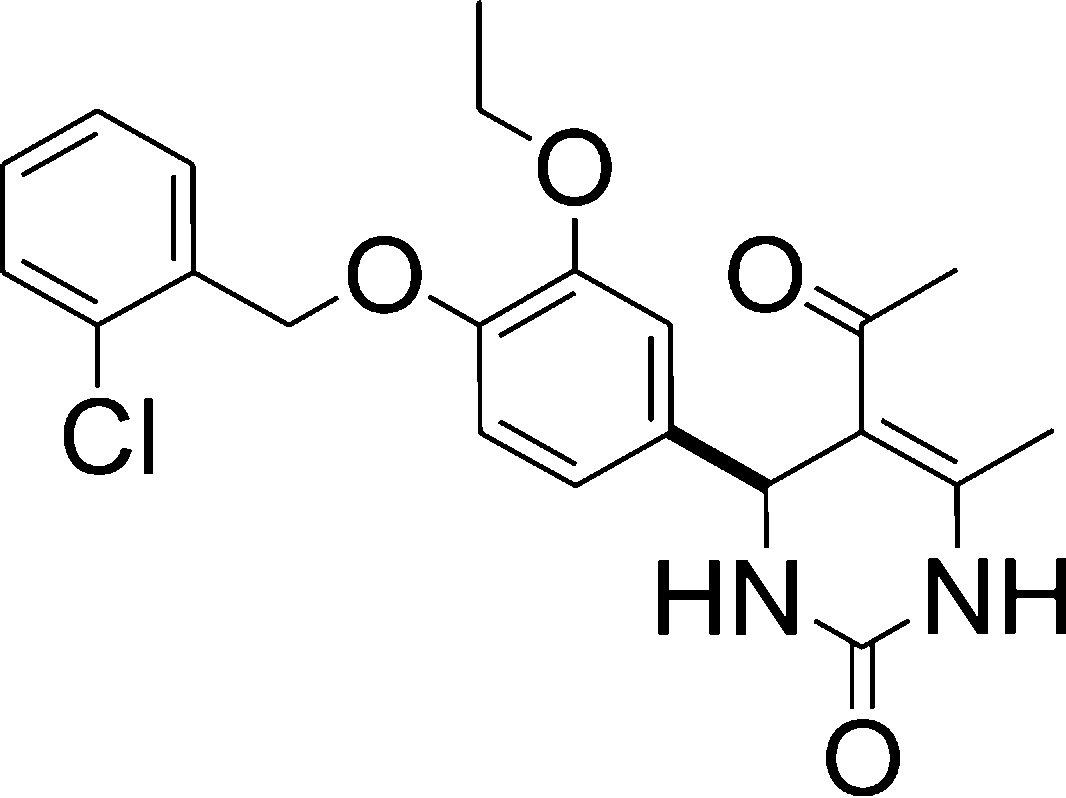
Compounds 1 and 4–39 (see Tables 1–4) were either commercially available or prepared by a Biginelli multicomponent reaction (Scheme 1) from appropriate 4-benzyloxybenzaldehydes I, beta-keto/esters/amides/ketones II, and (thio)ureas III, according to procedure A or B.15,16 When necessary, additional synthetic steps were performed to prepare compounds complementary to our pyrimidinone library.
Table 1. Evaluation of the Chirality Effect on EC50 and Emax.
| entry | compd | EC50 (μM) | Emax (% 7DC) |
|---|---|---|---|
| 1 | 1 (racemate) | 12.7 | 88 |
| 2 | 2 (enantiomer 1) | 10.9 | 85 |
| 3 | 3 (enantiomer 2) | >30a | >41b |
Not calculated (no plateau).
Not determined (no plateau).
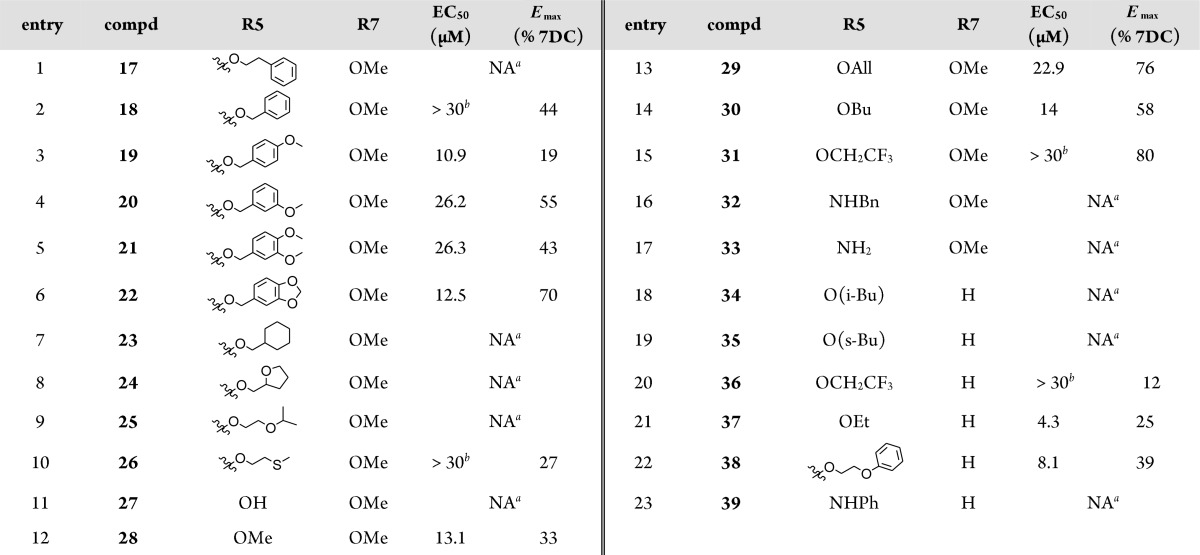
Table 4. SAR Elements Evidenced for the 5-Carbonyl Substituent.
NA: not active at 30 μM.
Not calculated (no plateau).
Two crystallographic structures of RORα are available in the Protein Data Bank (PDB). These structures are complexes of RORα with either the cholesterol (1N83)8 or the cholesterol sulfate (1SOX)9 ligands. They are considered to represent the active, agonist-induced conformation of the receptor. Superposition of the two structures showed that the main difference was located in the mobile loop between the alpha helices H1 and H2, whereas no differences were identified with respect to the positions that interact with the steroid ligands. We used these two structures as structural models for the docking experiments.
Although cholesterol forms an important hydrogen bond network with the protein through multiple water molecules, the sulfate group of the cholesterol sulfate replaces the water molecules to form direct hydrogen bonds with the residues of the mobile loop of the protein. These direct interactions modify the position of the mobile loop residues and consequently reduce the space volume of the entrance of the binding site of the protein. It is noteworthy that one water molecule is strictly conserved in the binding site between the two crystallographic complexes. This water molecule, located between alanine 330 and arginine 367 plays an important role in the hydrogen bond network for the binding of the two ligands.
Glide docking software was used in order to construct the model.17 Four models (the two crystallographic structures with or without the conserved water molecule) were evaluated for the docking procedure using the SP mode of Glide. The model formed by the protein part of the 1N83 structure and the conserved water molecule reproduced best the binding modes of both cholesterol and cholesterol sulfate. This model was used for all the experiments described in the following part.
Compounds were tested in dose response studies in the pull-down assay (Tables 1–4). As partial agonists, they can be differentiated both in terms of the affinity (EC50) and the efficacy (maximum effect, Emax). The affinity was used as the metrics to score compounds among them. The efficacy was used to highlight SAR elements or to differentiate compounds with comparable EC50 values.
Since Scaffold 1 contains an asymmetric center at C-4, the influence of its configuration on compound affinity was first considered. Enantiomers 2 and 3 (Table 1) were isolated from racemic compound 1 by semipreparative HPLC chiral chromatography and tested on the pull down assay. As expected, one enantiomer (compound 3) that was isolated in a high enantiomeric purity (96.78%) was found to be poorly potent, while enantiomer 2, isolated in a moderate enantiomeric purity (75.93%), accounted for most of the affinity of racemate 1
Docking studies with several potent scaffold 1 representatives were also performed to investigate the differences in the RORα binding mode of (S) and (R) enantiomers. Only (S) enantiomers had a reproducible and stable binding mode. Although these observations go along with the results obtained for compounds 2 and 3 in the pull-down assay, no specific configuration can be assigned to either compounds 2 and 3 without any dedicated analytical study (X-rays).
All further docking studies were carried on using the (S) configuration. However, compounds were still purchased or synthesized as racemates to rapidly produce structure–activity data.
We next examined the importance of the 4-benzyloxy group (Table 2, entries 1–10). While benzyl derivative 8 exhibited moderate affinity and efficacy, both the hydroxyl (compd 4), methoxy (compds 5 and 7) or propyloxy (compd 6) analogues were found to be not active. Moreover, chain lengthening from methylene (compd 9) to ethylene (compd 10) or propylene (compd 11) resulted in a loss of activity, both in terms of affinity and efficacy. Substitution of the benzyl ring also had beneficial effects; (4-(2-chloro)phenyl) derivative 13 was found active, while its non-halogenated analogue 12 was not.
Table 2. SAR Elements Evidenced for the Benzyloxy Group.
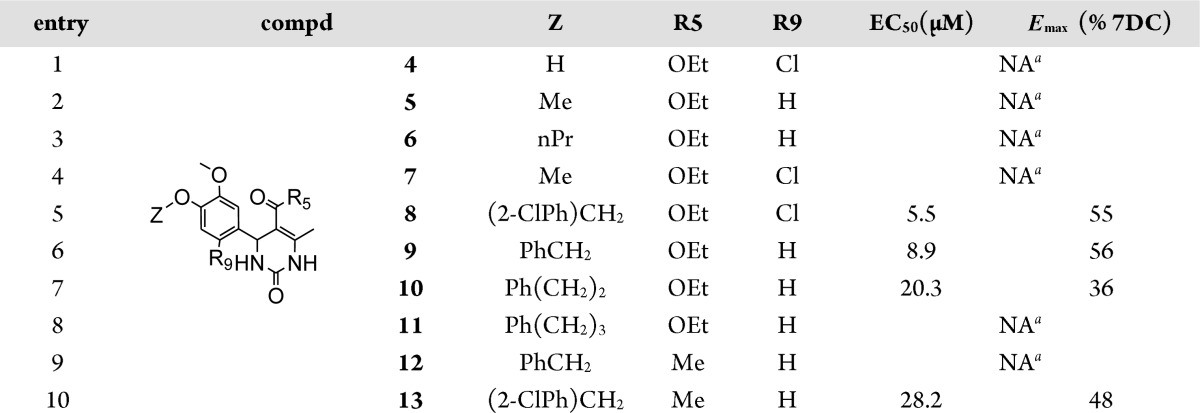
NA: not active at 30 μM.
The predicted binding mode found for the S-isomer of series best representative compound, compound 8, is shown in Figure 1. Interestingly, the aryl part is placed in the hydrophobic subpocket of the active site making it a privileged candidate for further optimization.
Figure 1.
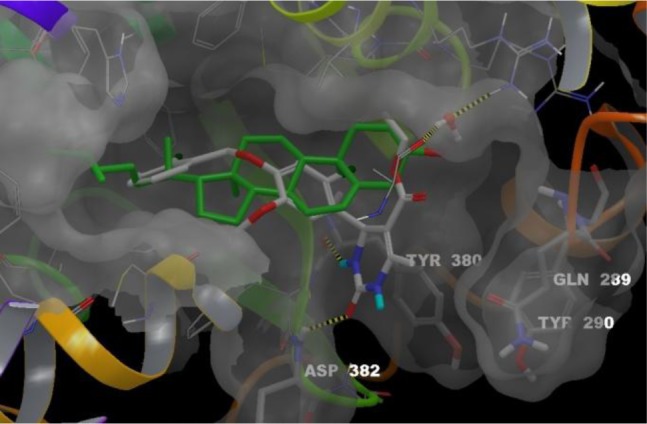
Superposition of cholesterol binding mode with compound 8-(S) predicted binding mode. Ligands, cholesterol (green carbons) and 8-(S) (white carbons), are colored by atom type. The conserved water molecule appears as balls and sticks. Four residues from RORα binding site are labeled in stick representation: Gln289 and Tyr290 materialize the mobile loop; Tyr380 and Asp382 that interact with 8-(S) through two hydrogen bonds (yellow dashed lines). These bonds between the NH in N-3 position and the backbone of Tyr380 and between the C=O of the urea moiety and the backbone of Asp382 anchor 8-(S) in the RORα binding site. Residues 327–335 (front yellow helix) are hidden.
SAR elements were also established for several positions of the dehydropyrimidinyl ring.
As observed in the pull-down assay, methylation of the nitrogen in the N-3 position (Table 3, R3 = Me) disrupted the activity of compound 15 as compared to compound 9, which is consistent with the binding mode reported in Figure 1. Methylation of the nitrogen in the N-1 position (compd 14) was also deleterious to compound affinity and efficacy as compared to compound 9.
Table 3. SAR Elements Evidenced for Positions 1, 3, and 6 of the Dehydropyrimidinyl Moiety.
| entry | compd | R1 | R3 | R6 | EC50 (μM) | Emax (% 7DC) |
|---|---|---|---|---|---|---|
| 1 | 14 | Me | H | Me | >19b | 29 |
| 2 | 15 | H | Me | Me | >30a | 19 |
| 3 | 16 | H | H | Et | >30a | >45b |
Not calculated (no plateau).
Not determined (no plateau).
In the C-6 position, replacement of the methyl (compd 9) by an ethyl (compd 16) led to a decrease in affinity and efficacy.
In the C-5 position, a panel of 6-methyl-2-oxo-5-substituted-1,2,3,4-tetrahydro-4-pyrimidinyl derivatives (Tables 2–4) has been tested so far. Among these, ethyl esters appeared as the best compromise between affinity and activity. Ethyl esters 8, 9, and 37 exhibited better affinities than bulkier esters 17, 18, 23–26, 29–31, 34, and 35. Similarly, ethyl ester 9 was more potent and efficacious than its methoxy analogue 28 or than hydrophilic acid and amide analogues 27 and 33. Other bulky analogues such as amide derivatives (compds 32 and 39) were inactive. Esters 19–21 were low-affinity and/or low-efficacy compounds as compared to compound 9.
However, bulky ester 38 was surprisingly as potent as compound 9, though less efficacious. Similarly, compound 22, a cyclic analogue of compounds 19–21, was more efficacious than compound 9, though less potent.
According to the proposed binding mode of compound 8-(S), the ester group in the C-5 position of the pyrimidinyl moiety is in proximity with the mobile loop (hydrophilic subpocket). Depending on the nature of the substituent in this position, water molecules can create bridges between the ligand and the loop, adapting this cavity to tolerate small to bulky groups. So far, the docking studies accuracy of 5-substituted-pyrimidinyl derivatives suffered from a relative lack of examples to properly model and anticipate their interactions with the mobile loop and also from the weakness of the docking software to efficiently take into account the high mobility of some parts of the receptors associated with the dynamic creation of water bridges.
Synthesis and testing of many more analogues will be helpful for the optimization process of the substituents in this particular position.
Finally, when comparing compounds 36 and 37, with compounds 31 and 9, respectively, it appeared that 3-methoxy substitution on the central phenyl ring favored the efficacy. A similar observation was made when comparing the compound 13 with 1 for which the 3-ethoxy substitution enhanced the efficacy.
In this study, various nonsteroidal RORα ligands (singletons or members of a hit series) were identified by a high throughput screening. As a result of the hit series evaluation process, 4-(4-(benzyloxy)phenyl)-5-carbonyl-2-oxo-1,2,3,4-tetrahydropyrimidine hit series was selected for further structure–activity relationship studies. Several additional compounds with moderate affinities and efficacies were identified, including the best compounds 8 and 9. Mostly, key structural elements were evidenced such as the benzyl and urea moieties, very important to maintain the activity or modifications on the central phenyl that enhance the efficacy. Moreover, these structure–activity results were consistent with the binding mode of the compound 8 (S-isomer) in the RORα docking model that was developed. Key hydrogen bonds between the urea moiety and the backbone of Tyr380 and Asp382 were identified. The benzyl group was localized in a wide hydrophobic pocket.
SAR knowledge reported herein will help to advance this chemical series further, to produce more potent RORα agonist compounds, notably through the optimization of both aromatic moieties. Preliminary evaluation has also shown that metabolic stability of this compound series needs to be improved. Our next goal is the conception of improved compounds, amenable to in vivo target validation studies.
Acknowledgments
We thank A.L. Enjalbert for the HPLC chiral analysis and the resolution methods setup and G. Bouly for performing the HPLC chiral semipreparative purifications.
Glossary
Abbreviations
- BAP
bacterial alkaline phosphatase
- GST
glutathione-S-transferase domain that binds glutathione
- LBD
ligand binding domain
- LXXLL
pentapeptide motif on coactivator proteins, required for an efficient nuclear receptor binding
- NR1F1, RORα
retinoic acid-related orphan receptor alpha
- TIF2 or NCOA2
nuclear receptor COActivator 2 protein
Supporting Information Available
Materials, full experimental procedures, and characterization of synthesized compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Screening campaign and hit series selection were performed at Genfit by S.H., S.D., M.B., M.D., and J. F.D. under the supervision of R.W. SAR and drug design were established and driven by M.D. Chemical synthesis was performed at ICOA by N.D., C.B., L.M., and M.D. under the supervision of S.R. The synthetic routes setup were realized by N.D. and C.B. Molecular modeling was performed at ICOA by L.C. under the supervision of L.M.-A. L.M., C.B., L.C., S.R., M.D., and R.W. authored the manuscript.
This research was accomplished as a part of the AD-Inov Program and supported by a grant from the French Fond Unique Interministériel (FUI).
The authors declare no competing financial interest.
Supplementary Material
References
- Hamilton B. A.; Frankel W. N.; Kerrebrock A. W.; Hawkins T. L.; et al. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature 1996, 379, 736–739. [DOI] [PubMed] [Google Scholar]
- Sidman R. L.; Lane P. W.; Dickie M. M. Staggerer, a new mutation in the mouse affecting the cerebellum. Science 1962, 137, 610–612. [DOI] [PubMed] [Google Scholar]
- Steinmayr M.; Andre E.; Conquet F.; Rondi-Reig L.; et al. Staggerer phenotype in retinoid-related orphan receptor alpha-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 3960–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duez H.; Staels B. The nuclear receptors Rev-erbs and RORs integrate circadian rhythms and metabolism. Diabetes Vasc. Dis. Res. 2008, 5, 82–88. [DOI] [PubMed] [Google Scholar]
- Fitzsimmons R. L.; Lau P.; Muscat G. E. , Retinoid-related orphan receptor alpha and the regulation of lipid homeostasis. J. Steroid Biochem. Mol. Biol. 2012, 130, 159–168. [DOI] [PubMed] [Google Scholar]
- Jetten A. M. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signaling 2009, 7, e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly S.; Journiac N.; Vernet-der Garabedian B.; Mariani J. RORalpha, a key to the development and functioning of the brain. Cerebellum 2012, 11, 451–452. [DOI] [PubMed] [Google Scholar]
- Kallen J. A.; Schlaeppi J. M.; Bitsch F.; Geisse S.; et al. X-ray structure of the hRORalpha LBD at 1.63 A: structural and functional data that cholesterol or a cholesterol derivative is the natural ligand of RORalpha. Structure 2002, 10, 1697–1707. [DOI] [PubMed] [Google Scholar]
- Kallen J.; Schlaeppi J. M.; Bitsch F.; Delhon I.; et al. Crystal structure of the human RORalpha Ligand binding domain in complex with cholesterol sulfate at 2.2 A. J. Biol. Chem. 2004, 279, 14033–14038. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Kumar N.; Crumbley C.; Griffin P. R.; et al. A second class of nuclear receptors for oxysterols: Regulation of RORalpha and RORgamma activity by 24S-hydroxycholesterol (cerebrosterol). Biochim. Biophys. Acta 2010, 1801, 917–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Kumar N.; Solt L. A.; Richardson T. I.; et al. Modulation of retinoic acid receptor-related orphan receptor alpha and gamma activity by 7-oxygenated sterol ligands. J. Biol. Chem. 2010, 285, 5013–5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.; Kojetin D. J.; Solt L. A.; Kumar K. G.; et al. Identification of SR3335 (ML-176): a synthetic RORalpha selective inverse agonist. ACS Chem. Biol. 2011, 6, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Kumar N.; Nuhant P.; Cameron M. D.; et al. Identification of SR1078, a synthetic agonist for the orphan nuclear receptors RORalpha and RORgamma. ACS Chem. Biol. 2010, 5, 1029–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra A. R.; Louet J. F.; Saha P.; An J.; et al. Absence of the SRC-2 coactivator results in a glycogenopathy resembling Von Gierke’s disease. Science 2008, 322, 1395–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R.; Mittal A.; Ramachandran U. Design and synthesis of 6-methyl-2-oxo-1,2,3,4-tetrahydro-pyrimidine-5-carboxylic acid derivatives as PPARγ activators. Bioorg. Med. Chem. Lett. 2007, 17, 4613–4618. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Wang Y.-Q.; Ge Z.-M.; Cheng T.-M.; et al. A highly efficient solvent-free synthesis of dihydropyrimidinones catalyzed by zinc chloride. Synthesis 2004, 1047–1051. [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.