

Abstract
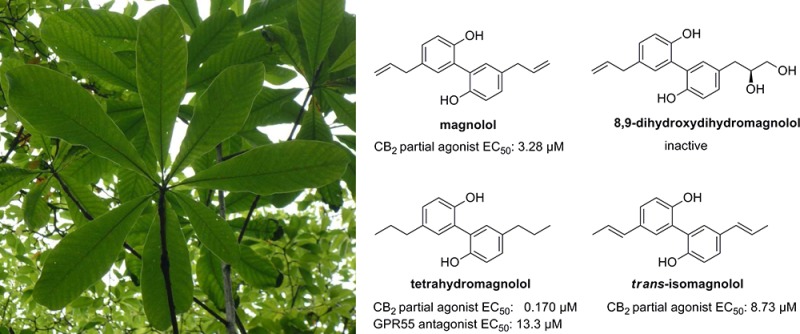
The bark of Magnolia officinalis is used in Asian traditional medicine for the treatment of anxiety, sleeping disorders, and allergic diseases. We found that the extract and its main bioactive constituents, magnolol and honokiol, can activate cannabinoid (CB) receptors. In cAMP accumulation studies, magnolol behaved as a partial agonist (EC50 = 3.28 μM) with selectivity for the CB2 subtype, while honokiol was less potent showing full agonistic activity at CB1 and antagonistic properties at CB2. We subsequently synthesized the major metabolites of magnolol and found that tetrahydromagnolol (7) was 19-fold more potent than magnolol (EC50 CB2 = 0.170 μM) exhibiting high selectivity versus CB1. Additionally, 7 behaved as an antagonist at GPR55, a CB-related orphan receptor (KB = 13.3 μM, β-arrestin translocation assay). Magnolol and its metabolites may contribute to the biological activities of Magnolia extract via the observed mechanisms of action. Furthermore, the biphenylic compound magnolol provides a simple novel lead structure for the development of agonists for CB receptors and antagonists for the related GPR55.
Keywords: bioactivation, biphenyls, CB2 receptor agonists, Chinese traditional medicine, honokiol, magnolia extract, Magnolia officinalis, magnolol, magnolol metabolites
The bark of Magnolia officinalis plays an important role in traditional Chinese and Japanese herbal medicine for the treatment of anxiety, sleep-related problems, and allergic diseases.1,2 The pharmacological effects were proposed to be mainly mediated by the neolignans honokiol (1), 4-methoxyhonokiol (2), magnolol (3), and (R)-8,9-dihydroxydihydromagnolol (4).2−4 An intensive search for their molecular targets revealed an interaction of honokiol and magnolol with a variety of enzymes and receptors at micromolar concentrations.5−8
We observed that the reported in vivo effects of M. officinalis extracts and its constituents possess a striking analogy to the effects described for ligands acting at cannabinoid (CB) receptors (Figure 1).9 Furthermore, the neolignans share structural similarity with some highly potent synthetic CB receptor ligands, such as CP55,940 (5), but also plant-derived biaryl CBs.8,10,11
Figure 1.

Structures of biologically active neolignans and synthetic CB receptor ligands.
CB receptors belong to the G protein-coupled receptor (GPCR) superfamily and are divided into two distinct subtypes designated CB1 and CB2, both of which are linked to an inhibition of adenylate cyclase.12 CB1 activation mediates analgesia, stimulation of appetite, and euphoria, among other effects.13 CB2 receptor activation results in analgesic and antiinflammatory effects.9 A related GPCR is the orphan GPR55, which has been described to be activated by lysophosphatidylinositol (LPI).14 Despite its low amino acid identity to CB1 (13.5%) and CB2 (14.4%) receptors, the GPR55 is reported to interact with certain CBs.14 For instance, the CB receptor agonist CP55,940 (5) behaves as a GPR55 antagonist, while the CB1-selective inverse agonist rimonabant (6) acts as an agonist at GPR55 (see the Supporting Information).12,14 The receptor appears to be involved in cancer cell proliferation and migration, angiogenesis, regulation of bone mass, and the onset of neuropathic pain.14−16
We investigated an ethanolic (90%) extract of M. officinalis bark (magnolol content, 17.9%; honokiol, 22.8%; also see the Supporting Information) for its affinity to CB receptors. In addition, magnolol and honokiol, the major neolignans found in Magnolia bark (0.78–7.65 and 0.17–1.81%, respectively), were studied in radioligand binding and cAMP accumulation assays at CB1 and CB2 receptors and in β-arrestin translocation assays at the GPR55.
Magnolol is known to be extensively metabolized by tissue and intestinal bacterial enzymes to hydrogenated and hydroxy derivatives, glucuronides, and sulfates.2 The main fecal metabolites of orally administered magnolol (3) in rats are tetrahydromagnolol (7) and trans-isomagnolol (8).17 Interestingly, the amount of tetrahydromagnolol (7) formed had been found to be increased in rats after repeated administration of magnolol (3), indicating the involvement of inducible enzymes in the metabolization of magnolol (3).17,18 Because metabolites may contribute to the action of drugs, we additionally synthesized and investigated 7 and 8.
The synthesis of 7 by phenolic oxidative coupling had previously been described.19 In the present study, we report a new synthetic route to obtain 7 in a higher yield of 25% (Scheme 1A). 2-Bromo-4-propylphenol (10) was prepared starting from 4-propylphenol (9) by electrophilic aromatic substitution.20 Boronic acid (11) was obtained by treatment of 10 with butyllithium and subsequent reaction with trimethyl borate followed by acidic hydrolysis.21 The coupling of 10 with 11 was performed by a Suzuki–Miyaura cross-coupling reaction procedure, which had previously been used for the preparation of honokiol derivatives,22 affording the desired tetrahydromagnolol (7). trans-Isomagnolol (8) was obtained by a new procedure depicted in Scheme 1B. 2,2′-Dimethoxybiphenyl (12) was brominated with N-bromosuccinimide to give 5,5′-dibromo-2,2′-dimethoxybiphenol (13).23 After demethylation of 13 to 5,5′-dibromo-2,2′-diol (14)24 and subsequent protection of 14 to the acetylated intermediate 5,5′-dibromo-2,2′-diacetate (15), a Suzuki–Miyaura cross-coupling reaction was performed with trans-propenylboronic acid to yield 5,5′-di-((E)-prop-1-enyl)biphenyl-2,2′-diacetate (16).24 Product 8 was obtained by basic ester cleavage of 16.25 Another metabolite, primarily found in the human urine after oral administration of M. officinalis bark preparations, is 8,9-dihydroxydihydromagnolol (4). Compound 4 was synthesized as depicted in Scheme 1C by treatment of 3 with osmium tetroxide to afford 4 in 20% yield after purification.26
Scheme 1. Synthesis of Tetrahydromagnolol,trans-Isomagnolol, and 8,9-Dihydroxydihydromagnolol.

Reagents and conditions: (a) Br2, NaHCO3, CHCl3, 0 °C, 80% yield. (b) Three steps: (1) butyl lithium, Et2O, −78 °C; (2) B(OCH3)3, Et2O, −78 °C to rt; and (3) HCl, Et2O, 50% yield. (c) Pd(PPh3)4, Na2CO3, toluene, EtOH, H2O, 100 °C, 25% yield.
Reagents and conditions: (a) N-Bromosuccinimide, dimethylformamide, rt, 12 h, 80% yield. (b) CH2Cl2, BBr3, −78 °C to rt, 75% yield. (c) Acetic anhydride, 120 °C, 2 h, 95% yield. (d) trans-1-Propene-1-boronic acid, Pd(PPh3)4, CsF, dioxane, water, 85% yield. (e) NaHCO3, water, methanol, 90% yield.
Reagents and conditions: (a) OsO4, t-BuOH, acetone, water, 24 h, 20%.
The ethanolic M. officinalis bark extract (“M-extract”) showed high affinity in radioligand binding assays for human CB1 and CB2 receptor subtypes recombinantly expressed in Chinese hamster ovary (CHO-K1) cells (for assay procedures, see the references).27,28 As depicted in Figure 2, the extract preferentially displaced the radioligand at CB2 receptors (Ki = 0.165 μg/mL at CB2, 1.21 μg/mL at CB1; >7-fold difference).
Figure 2.
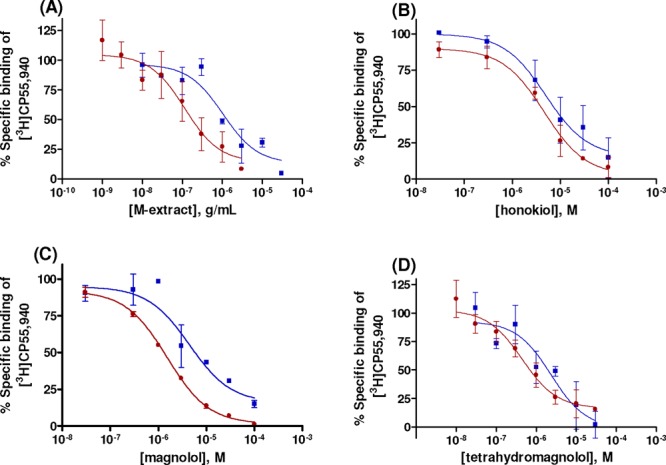
Concentration-dependent inhibition of specific [3H]CP55,940 binding at membrane preparations of CHO cells recombinantly expressing human CB1 (blue square) or CB2 (maroon circle) receptors by (A) magnolia extract, (B) honokiol (1), (C) magnolol (3), and (D) tetrahydromagnolol (7). Data points represent means ± SEMs of three independent experiments performed in duplicates.
Magnolol, but not honokiol, also appeared to show a certain preference for CB2 in binding studies (Ki values of magnolol at CB1, 3.19 μM; at CB2, 1.44 μM; honokiol at CB1, 6.46 μM; at CB2, 5.61 μM; Figure 2B,C and Table 1). In cAMP assays, the difference was even more pronounced, magnolol showing a 6-fold higher potency at CB2 as compared to CB1 receptors (Table 1 and Figure 3A).
Table 1. Interaction of Magnolia Extract and Test Compounds with CB1 and CB2 Receptors and GPR55.
| radioligand
binding vs
[3H]CP55,940 |
cAMP accumulation assay |
β-arrestin assay | |||
|---|---|---|---|---|---|
|
Ki ± SEM (μM), n = 3 |
EC50 ± SEM (μM), n = 3 [efficacy at 100 μM
as compared to max effect of full agonist 5 (1 μM)
= 100%] |
KB ± SEM (μM), n = 3 (inihibition of LPI-induced β-arrestin translocation at 10 μM) | |||
| compd | human CB1 receptor | human CB2 receptor | human CB1 receptor | human CB2 receptor | human GPR55 |
| honokiol (1) | 6.46 ± 3.54 | 5.61 ± 2.02 | >10.0 (168%) | NDa (0%) | NDa (42%)b |
| magnolol (3) | 3.15 ± 1.65 | 1.44 ± 0.10 | 18.3 ± 8.6 (62%) | 3.28 ± 2.01 (31%)c | NDa (0%)b |
| (RS)-8,9-dihydroxydihydro-magnolol (4) | >10.0 | >10.0 | NDa | NDa | NDa (4%)b |
| tetrahydro-magnolol (7) | 2.26 ± 0.89 | 0.416 ± 0.089 | 9.01 ± 3.42 (124%) | 0.170 ± 0.114 (49%)d | 13.3 ± 2.0 (96%)b |
| trans-isomagnolol (8) | >10.0 | 3.14 ± 0.12 | >10.0 (134%) | 8.73 ± 3.39 (55%) | NDb (0%)b |
| CP55,940 (5) | 0.00128 ± 0.00044 | 0.00142 ± 0.00075 | 0.00228 ± 0.00137c | 0.00100 ± 0.00019 | 1.89 ± 0.97 (92%)b |
| rimonabant (6) | 0.0126 ± 0.0039 | 0.900 ± 0.320 | NDa (0%)c,d | NDa (0%)d | NDa (agonist: EC50 = 2.01 ± 0.66) |
| magnolia extract | 1.21 ± 0.48 μg/mL | 0.165 ± 0.104 μg/mL | NDa (147%)e | NDa (0%)e | NDa (121%)b |
ND, not determined.
Inhibition of LPI (1 μM)-induced β-arrestin recruitment by test compounds (at 10 μM; 50 μg/mL for magnolia extract). The LPI effect is set at 100%.
n = 4.
Concentration of test compound (10 μM).
Concentration of magnolia extract (50 μg/mL).
Figure 3.
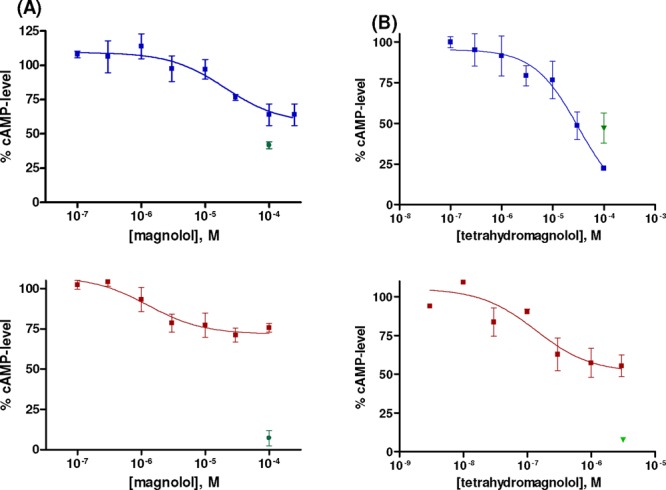
Concentration-dependent inhibition of forskolin (10 μM)-induced cAMP accumulation in CHO cells recombinantly expressing the human CB1 (blue square) or CB2 (maroon square) receptor by (A) magnolol (3) and (B) tetrahydromagnolol (7). The effect of the full agonist CP55,940 (1 μM) is depicted by a green symbol (green upside down triangle). Data points represent means ± SEMs of at least three independent experiments each performed in duplicates.
The functional studies indicated that magnolol (3) is a partial agonist at both receptor subtypes in comparison to the full agonist 5. Although 3 possessed higher potency at CB2 as compared to CB1, it exhibited lower efficacy (CB1, 62%; CB2, 31%; Table 1 and Figure 3). In contrast, honokiol and the M-extract showed full agonistic properties at CB1 receptors, while they acted as antagonists with inverse agonistic activity at the CB2 receptor subtype (Table 1 and Supporting Information).
In radioligand binding experiments as well as in functional studies, the main fecal magnolol metabolite 7 showed a similar profile as the parent natural product 3 but was considerably more potent (Table 1 and Figure 2). Like magnolol (3), metabolite 7 was CB2-selective, being a partial agonist at CB2 receptors and a full agonist at CB1. In cAMP assays at CB2 receptors, 7 (EC50 = 0.170 μM) was almost 20-fold more potent than 3 (Figure 3B). These findings indicate that the modestly potent magnolol (3) is bioactivated to the potent metabolite 7 and can thus be envisaged as a pro- or codrug. The second fecal magnolol metabolite, 8, was also found to be active at CB receptors and displayed comparable functional properties as 3 and 7 but possessed lower affinity and potency at both receptor subtypes (see Table 1 and Supporting Information). In contrast to tetrahydromagnolol (7) and trans-isomagnolol (8), the main urinary metabolite of magnolol in humans, 8,9-dihydroxydihydromagnolol (4), showed no affinity for CB receptors, indicating that its described antidepressant effects are likely not mediated by CB receptors (Table 1).2
As a next step, the magnolia extract and the individual compounds were investigated for their potential to interact with the CB-like orphan receptor GPR55 by performing β-arrestin translocation assays. The M-extract, tested at a concentration of 50 μg/mL, showed complete inhibition of the LPI effect (Table 1), indicating that in addition to CB receptors, interaction with the GPR55 may also be involved in mediating the biological effects of M. officinalis extracts. As depicted in Figure 4A, none of the pure biphenyls was able to significantly induce β-arrestin recruitment via GPR55 activation at a high concentration of 10 μM. This indicated that they did not act as GPR55 agonists.
Figure 4.

(A) Effects of LPI (GPR55 agonist, 1 μM), honokiol (1), magnolol (3), 8,9-dihydroxydihydromagnolol (4), tetrahydromagnolol (7), and trans-isomagnolol (8) (10 μM each) on β-arrestin recruitment to human GPR55 receptors recombinantly expressed in CHO cells. (B) Inhibition of LPI (1 μM)-induced β-arrestin recruitment to human GPR55 by 1, 3, 4, 7, and 8 (10 μM). The results in A and B represent means ± SEMs of three independent experiments performed in duplicates. Data are expressed as the percent luminescence related to the effect of LPI (1 μM; ≥50% of maximal effect; EC50 = 0.769 μM) set at 100%. (C) Concentration-dependent effect of LPI on β-arrestin recruitment to human GPR55 receptors recombinantly expressed in CHO cells in the absence and in the presence of tetrahydromagnolol (7, 10 μM). Data points are means ± SEMs of three independent experiments, performed in duplicates. A KB value of 13.3 ± 2.0 μM was determined for tetrahydromagnolol; RLU = relative luminescence units. Recruitment of β-arrestin to the receptor was detected by measuring luminescence emission, based on a β-galactosidase enzyme fragment complementation assay (DiscoverX).
However, when tested for potential inhibitory effects, honokiol (1) and—more strongly—tetrahydromagnolol (7) were able to inhibit LPI-induced GPR55 activation at a concentration of 10 μM (Figure 4B), while magnolol (3), 8,9-dihydromagnolol (4), and trans-isomagnolol (8) were inactive. The more potent 7 was further investigated for its antagonistic properties at GPR55. Tetrahydromagnolol (7, 10 μM) led to a significant parallel rightward shift of the curve for the agonist LPI at GPR55, indicating a competitive mechanism of inhibition. A KB value of 13.3 μM was determined (Figure 4C).
The magnolia extract and biphenyls 1, 3, 4, 7, and 8 were further investigated for potential inhibition of the endocannabinoid-degrading enzymes fatty acid amide hydrolase (FAAH) and monoacylglyceride lipase (MAGL) in rat brain preparations.29,30 No inhibition of the enzymes was observed by the compounds at a test concentration of 10 μM. The extract, tested at a high concentration of 50 μg/mL, did not inhibit FAAH and showed only weak inhibition (22%) of MAGL (for details, see the Supporting Information).
During the preparation of this manuscript, a study appeared describing honokiol, 4-methoxyhonokiol, and magnolol as CB2 receptor ligands.8 4-Methoxyhonokiol was found to exhibit high affinity (44 nM), while magnolol and honokiol were described to possess moderate (1–3 μM) affinities for CB2 receptors as determined in radioligand binding studies.8 These findings are in accordance with the results obtained in our laboratory. Furthermore, it was reported that magnolol and honokiol possess a significantly lower affinity for the CB1 receptor.8 In our hands, this observation was only true for magnolol, while honokiol possessed about equal affinity for both CB receptor subtypes.
No functional characterization of the main neolignan constituents of Magnolia bark, magnolol, and honokiol was described in the recently published study,8 neither at CB1 nor at CB2 receptors. In the present study, we describe for the first time the functional properties of the natural products honokiol (CB1 agonist and CB2 antagonist) and magnolol (partial agonist at both receptors with preference for CB2).
Moreover, we discovered that its main metabolites trans-isomagnolol and tetrahydromagnolol are selective partial CB2 agonists. The main metabolite of magnolol, tetrahydromagnolol, is in fact a more potent partial CB2 agonist than the parent drug magnolol and also shows CB2 selectivity. Partial CB1 and CB2 receptor agonists had been shown to reduce inflammation-associated hyperalgesia and to possess antiallodynic effects in animal models.31,32 Thus, magnolol and its metabolites may have the same in vivo effects due to their action on CB receptors.
Furthermore, magnolia extract and its constituents magnolol and honokiol, as well as the main metabolites of magnolol 4, 7, and 8, were investigated for the first time at the CB-like GPR55 using β-arrestin translocation assays and at enzymes responsible for endocannabinoid degradation. Compound 7 was found to be a moderately potent GPR55 antagonist (KB = 13.3 μM). It could serve as a lead structure for the development of potent and selective GPR55 antagonists.
Our results together with recently published reports provide a new potential mechanism of action for the known effects of Magnolia bark extracts. An involvement of further bioactive constituents, as well as synergistic effects, have to be considered. As blood levels of 7 after magnolol intake have not been determined yet, pharmacokinetic studies are needed to corroborate our findings. Metabolite 7 exhibits an approximately 11-fold lower affinity for CB2 receptors as compared to the well-known plant-derived CB Δ9-THC. The lower affinity of 7 might be sufficient for biological activity, particularly since it may accumulate after repeated intake of magnolol-containing formulations.19,20 Further investigations using in vivo models are warranted. The biphenyl magnolol provides a simple, novel lead structure for the development of agonists for CB receptors and antagonists for the related GPR55.
Acknowledgments
We thank Walburga Hanekamp for technical assistance.
Supporting Information Available
Synthetic procedures, analytical data, and assay procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Status
∥ T.K. was on leave from the Jagiellonian University, Cracow, Poland.
Supplementary Material
References
- Koetter U.; Barrett M.; Lacher S.; Abdelrahman A.; Dolnick D. Interactions of Magnolia and Ziziphus extracts with selected central nervous system receptors. J. Ethnopharmacol. 2009, 124, 421–425. [DOI] [PubMed] [Google Scholar]
- Lee Y. J.; Lee Y. M.; Lee C. K.; Jung J. K.; Han S. B.; Hong J. T. Therapeutic applications of compounds in the Magnolia family. Pharmacol. Ther. 2011, 130, 157–176. [DOI] [PubMed] [Google Scholar]
- Jada S.; Reddy Doma M.; Singh P. P.; Kumar S.; Malik F.; Sharma A.; Khan I. A.; Qazi G. N.; Kumar H. M. Design and synthesis of novel magnolol derivatives as potential antimicrobial and antiproliferative compounds. Eur. J. Med. Chem. 2012, 51, 35–41. [DOI] [PubMed] [Google Scholar]
- Steinmann P.; Walters D. K.; Arlt M. J.; Banke I. J.; Ziegler U.; Langsam B.; Arbiser J.; Muff R.; Born W.; Fuchs B. Antimetastatic activity of honokiol in osteosarcoma. Cancer 2012, 118, 2117–2127. [DOI] [PubMed] [Google Scholar]
- Schuehly W.; Khan S. I.; Fischer N. H. Neolignans from North American Magnolia species with cyclooxygenase 2 inhibitory activity. Inflammopharmacology 2009, 17, 106–110. [DOI] [PubMed] [Google Scholar]
- Schuehly W.; Paredes J. M.; Kleyer J.; Huefner A.; Anavi-Goffer S.; Raduner S.; Altmann K. H.; Gertsch J. Mechanisms of osteoclastogenesis inhibition by a novel class of biphenyl-type cannabinoid CB2 receptor inverse agonists. Chem. Biol. 2011, 18, 1053–1064. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Xu X.; Chen L.; Chen J.; Hu L.; Jiang H.; Shen X. Molecular determinants of magnolol targeting both RXRalpha and PPARgamma. PLoS One 2011, 6, e28253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taferner B.; Schuehly W.; Huefner A.; Baburin I.; Wiesner K.; Ecker G. F.; Hering S. Modulation of GABAA-receptors by honokiol and derivatives: subtype selectivity and structure-activity relationship. J. Med. Chem. 2011, 54, 5349–5361. [DOI] [PubMed] [Google Scholar]
- Pacher P.; Batkai S.; Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M.; Kinugawa T.; Asakawa Y. Bibenzyl cannabinoid and bisbibenzyl derivative from the liverwort Radula perrottetii. Phytochemistry 1994, 37, 859–862. [Google Scholar]
- Worm K.; Zhou Q. J.; Stabley G. J.; DeHaven R. N.; Dolle R. E. Biaryl cannabinoid mimetics-synthesis and structure-activity relationship. Bioorg. Med. Chem. Lett. 2007, 17, 3652–3656. [DOI] [PubMed] [Google Scholar]
- Kapur A.; Zhao P.; Sharir H.; Bai Y.; Caron M. G.; Barak L. S.; Abood M. E. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J. Biol. Chem. 2009, 284, 29817–29827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger S.; Nickl K.; Schneider E. H.; Seifert R.; Heilmann J. Establishment of recombinant cannabinoid receptor assays and characterization of several natural and synthetic ligands. Naunyn Schmiedebergs Arch. Pharmacol. 2010, 382, 177–191. [DOI] [PubMed] [Google Scholar]
- Sharir H.; Abood M. E. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol. Ther. 2010, 126, 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henstridge C. M.; Balenga N. A.; Schröder R.; Kargl J. K.; Platzer W.; Martini L.; Arthur S.; Penman J.; Whistler J. L.; Kostenis E.; Waldhoer M.; Irving A. J. GPR55 ligands promote receptor coupling to multiple signalling pathways. Br. J. Pharmacol. 2010, 160, 604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. A. L-alpha-lysophosphatidylinositol meets GPR55: A deadly relationship. Trends Pharmacol. Sci. 2011, 32, 265–269. [DOI] [PubMed] [Google Scholar]
- Hattori M.; Sakamoto T.; Endo Y.; Kakiuchi N.; Kobashi K.; Mizuno T.; Namba T. Metabolism of magnolol from magnoliae cortex. I. Application of liquid chromatography-mass spectrometry to the analysis of metabolites of magnolol in rats. Chem. Pharm. Bull. (Tokyo) 1984, 32, 5010–5017. [DOI] [PubMed] [Google Scholar]
- Hattori M.; Endo Y.; Takebe S.; Kobashi K.; Fukasaku N.; Namba T. Metabolism of magnolol from Magnoliae cortex. II. Absorption, metabolism and excretion of [ring-14C]magnolol in rats. Chem. Pharm. Bull. (Tokyo) 1986, 34, 158–167. [DOI] [PubMed] [Google Scholar]
- Kong Z. L.; Tzeng S. C.; Liu Y. C. Cytotoxic neolignans: an SAR study. Bioorg. Med. Chem. Lett. 2005, 15, 163–166. [DOI] [PubMed] [Google Scholar]
- Ulrich O.; Pfeiffer H. P.; Breitmaier E. Taschenporphyrine mit intramolekularen Liganden. Chem. Ber. 1986, 119, 3507–3514. [Google Scholar]
- Konakahara T.; Kiran Y. B.; Okuno Y.; Ikeda R.; Sakai N. An expedient synthesis of ellipticine via Suzuki-Miyaura coupling. Tetrahedron Lett. 2010, 51, 2335–2338. [Google Scholar]
- Suzuki A.; Miyaura N. A. New stereospecific cross-coupling by the palladium catalized reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 36, 3437–3440. [Google Scholar]
- Agharahimi M. R.; LeBel N. A. Synthesis of (−)-monoterpenylmagnolol and magnolol. J. Org. Chem. 1995, 60, 1856–1863. [Google Scholar]
- Crosignani S.; Pretre A.; Jorand-Lebrun C.; Fraboulet G.; Seenisamy J.; Augustine J. K.; Missotten M.; Humbert Y.; Cleva C.; Abla N.; Daff H.; Schott O.; Schneider M.; Burgat-Charvillon F.; Rivron D.; Hamernig I.; Arrighi J. F.; Gaudet M.; Zimmerli S. C.; Juillard P.; Johnson Z. Discovery of potent, selective, and orally bioavailable alkynylphenoxyacetic acid CRTH2 (DP2) receptor antagonists for the treatment of allergic inflammatory diseases. J. Med. Chem. 2011, 54, 7299–7317. [DOI] [PubMed] [Google Scholar]
- Büchi G.; Weinreb S. M. Total syntheses of aflatoxins M1 and G1 and an improved synthesis of aflatoxin B1. J. Am. Chem. Soc. 1940, 62, 1963–1967. [DOI] [PubMed] [Google Scholar]
- Van Rheenen V.; Kelly R. C.; Cha D. Y. An improved catalytic OsO4 oxidation of olefins to cis-1,2-glycols using tertiary amine oxides as the oxidant. Tetrahedron Lett. 1976, 25, 1973–1976. [Google Scholar]
- Behrenswerth A.; Volz N.; Torang J.; Hinz S.; Brase S.; Müller C. E. Synthesis and pharmacological evaluation of coumarin derivatives as cannabinoid receptor antagonists and inverse agonists. Bioorg. Med. Chem. 2009, 17, 2842–2851. [DOI] [PubMed] [Google Scholar]
- Elsebai M. F.; Rempel V.; Schnakenburg G.; Kehraus S.; Müller C. E.; König G. M. Identification of a potent and selective cannabinoid CB1 receptor antagonist from Auxarthron reticulatum. ACS Med. Chem. Lett. 2011, 2, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster L.; Schulze Elfringhoff A.; Lehr M. High-performance liquid chromatographic assay with fluorescence detection for the evaluation of inhibitors against fatty acid amide hydrolase. Anal. Bioanal. Chem. 2009, 394, 1679–1685. [DOI] [PubMed] [Google Scholar]
- Holtfrerich A.; Makharadze T.; Lehr M. High-performance liquid chromatography assay with fluorescence detection for the evaluation of inhibitors against human recombinant monoacylglycerol lipase. Anal. Biochem. 2010, 399, 218–224. [DOI] [PubMed] [Google Scholar]
- Pertwee R. G. The therapeutic potential of drugs that target cannabinoid receptors or modulate the tissue levels or actions of endocannabinoids. AAPS J. 2005, 7, E625–E654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vry J.; Denzer D.; Reissmueller E.; Eijckenboom M.; Heil M.; Meier H. Mauler, F. 3-[2-cyano-3-(trifluoromethyl) phenoxy]phenyl-4,4,4-trifluoro-1-butanesulfonate (BAY 59-3074): A novel cannabinoid CB1/CB2 receptor partial agonist with antihyperalgesic and antiallodynic effects. J. Pharmacol. Exp. Ther. 2004, 310, 620–632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.