

Abstract
TGR5 is a G-protein-coupled receptor (GPCR) mediating cellular responses to bile acids (BAs). Although some efforts have been devoted to generate homology models of TGR5 and draw structure–activity relationships of BAs, none of these studies has hitherto described how BAs bind to TGR5. Here, we present an integrated computational, chemical, and biological approach that has been instrumental to determine the binding mode of BAs to TGR5. As a result, key residues have been identified that are involved in mediating the binding of BAs to the receptor. Collectively, these results provide new hints to design potent and selective TGR5 agonists.
Keywords: Bile acids, TGR5, diabetes, GPCR, site-directed mutagenesis, homology modeling, molecular docking
Lipid receptors are a growing family of druggable targets that mediate a plethora of signaling pathways involved in the fine-tuning of important physiological functions such as the control of metabolism, organ physiology, cell differentiation, and homeostasis.1 Independently discovered by two Japanese research groups,2,3 TGR5 is a membrane lipid receptor G-protein coupled to the production of cAMP and activated by BAs (1-4). The activation of TGR5, in particular, bestows on BAs the ability to modulate nongenomic signaling pathways that complement their genomic actions, which are mostly mediated by the interaction with the nuclear receptor FXR.4 Major TGR5-dependent actions of BAs include immunosuppressive properties and the regulation of glucose metabolism as well as energy homeostasis.5−9 While the therapeutic relevance of the immune properties of TGR5 activation is pending further appraisals, the effect of the receptor on glucose metabolism and energy homeostasis have thrust TGR5 into the limelight as an attractive therapeutic target in the arena of metabolic disorders, including type-2 diabetes (T2D) and obesity.10−12
The past decade has witnessed an intense research activity on part of both academia and pharmaceutical companies toward the identification, design, and synthesis of potent and selective modulators of TGR5 for drug development.13−18 These efforts have combined with attempts to understand how ligands interact within the binding cleft of the receptor.19 Although no crystal structure of TGR5 is hitherto available, ligand-based approaches and homology modeling studies have been instrumental to depict features of the receptor binding site,20−24 which have been used to aid the development of new ligands25 and the interpretation of TGR5 polymorphisms in patients with primary sclerosing cholangitis.26 In this letter, we report the results of an integrated computational, biological, and chemical approach that has been designed to probe the so far elusive binding mode of BAs to TGR5. The starting point of the approach was the generation of a 3D-model of human TGR5 (Figure 1), using the inactive state of rhodopsin GPCR as template structure (pdb code: 1L9H).
Figure 1.
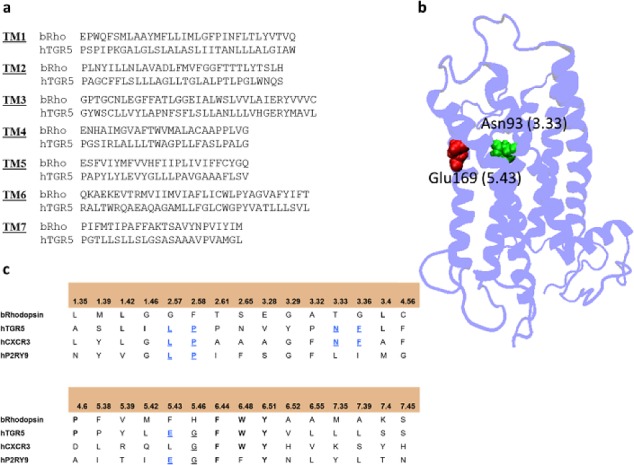
(a) Sequence alignment of bovine rhodopsin receptor and human TGR5; (b) 3D model of TGR5; (c) alignment of 30 binding site residues according to Surgand and co-workers.27
Although in one of previous works the structure of the adenosine A2A receptor was used as template for TGR5 modeling,23 we selected the rhodopsin structure as template on the basis of the following considerations: (a) the sequence of rhodopsin GPCR shares a good similarity with human TGR5 sequence (15% identities; 26% similarities); (b) both receptors have one disulfide bridge and feature a similar length of the second extracellular loop (EL2), which is thought to play a key role in GPCR activation. It should be mentioned that the rhodopsin structure was also used as a template to build a homology model of TGR5 in a more recent work.24 To identify next the putative BAs binding site in TGR5, we adopted the strategy developed by Surgand and co-workers.27 In particular, the authors identified residues occupying 30 discontinuous positions in TM helices that lined the binding pocket of retinal in the rhodopsin structure and clustered 369 nonredundant GPCRs on the basis of similarity to these residues. The resulting clusters were able to reproduce all known GPCR subfamilies, with most of the residues known to affect ligand binding in GPCRs being included in the list of 30 binding site residues. Accordingly, we identified two positions (3.33 and 5.43) from the inspection of TGR5 sequence alignment that featured residues conserved with as many residues in two GPCRs, namely, purinergic receptor P2Y9 and chemokine receptor CXC3, being involved in key ligand interactions (Figure 1).
Embracing the hypothesis that Asn93 in position 3.33 and Glu169 in position 5.53 were involved in defining the binding site of TGR5, a first round of mutagenesis experiments was designed, engineering TGR5Asn93Ala and TGR5Glu169Ala mutant receptors. The biological appraisal of LCA (1), CA (2), and CDCA (3) as well as semisynthetic BAs derivatives 4 and 5 showed that the activity of these compounds was abolished in TGR5Asn93Ala and reduced in TGR5Glu169Ala (Table 1), pinpointing a role for Asn93 and Glu169 in the activation of TGR5 by BAs. On the basis of this evidence, docking calculations were carried out on potent TGR5 agonists, such as S-EMCA (INT-777, 4) and 6-ECDCA (obeticholic acid, OCA, INT-747, 5) around Asn93 and Glu169 of the receptor binding site.
Table 1. Fold change in EC50 compared to TGR5WT.
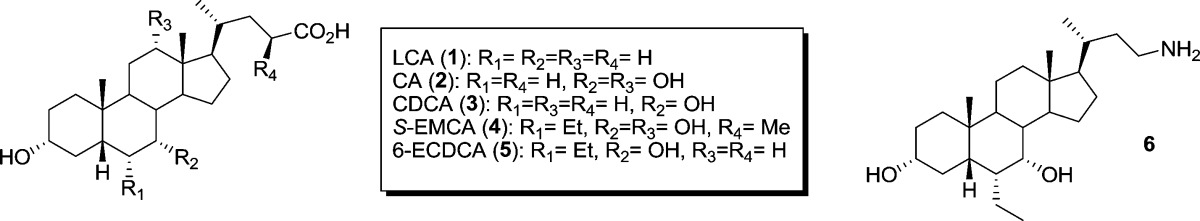
| name | WT | N93A | N93D | E169A | N76A | Y89A | Y89F | S270A |
|---|---|---|---|---|---|---|---|---|
| LCA (1) | 0.73 μM | >1000 | 39.1 | 0.6 | 13.9 | 1.10 | >1000 | |
| CA (2) | 19.83 μM | >1000 | 29.1 | 0.8 | 81.9 | 0.22 | >1000 | |
| CDCA (3) | 10.29 μM | >1000 | 69.5 | 1.0 | 88.4 | 0.18 | >1000 | |
| S-EMCA (4) | 0.18 μM | >1000 | 52.4 | 3.9 | >100 | 1.45 | >1000 | |
| ECDCA (5) | 0.10 μM | >1000 | 9.4 | 25.5 | 1.4 | >100 | 0.82 | >1000 |
| 6 | 1.32 μM | >1000 | >1000 | 1.70 | >1000 |
As a result, two distinct groups of docking poses were obtained for 4 and 5 (Tables S1–S2 of the Supporting Information), spreading in a window of energy scores (IFD score) of 5.2 and 4.5 kcal/mol from the top scored pose, respectively. The best docking pose in terms of binding score energy (4 IFD = −622.3 kcal/mol; 5 IFD score = −621.8 kcal/mol; binding mode 1) and the most diverse pose in terms of rmsd (4 rmsd = 8.84 Å; 5 rmsd = 8.35 Å; binding mode 2) were both selected for further analysis. These binding poses are schematically shown in Figure 2, namely, a tail-to-head pose (binding mode 1) and head-to-tail pose (binding mode 2). Both binding poses involved hydrogen-bonding interactions with Asn93, in agreement with results from the first round of mutagenesis experiments, and the side chain of Tyr89. Although no interactions were observed with Glu169, we envisaged a potential role for this residue in the transduction of signal rather than in the binding of BAs, given its remote location (∼12 Å) from the preliminary docked poses of BAs. An additional polar residue lining the binding pose of S-EMCA (4) and 6-ECDCA (5) was Asn76, albeit no hydrogen-bonding interaction was observed involving this residue. Nonpolar interactions were also observed between the steroid scaffold of BAs and the side chains of Leu68, Pro72, Pro92, Phe96, Trp237, and Leu266 (Figures S1–S2 of the Supporting Information). A second round of mutagenesis experiments was designed on the basis of initial docking results, engineering the following two mutant receptors: TGR5Tyr89Ala and TGR5Asn76Ala.
Figure 2.
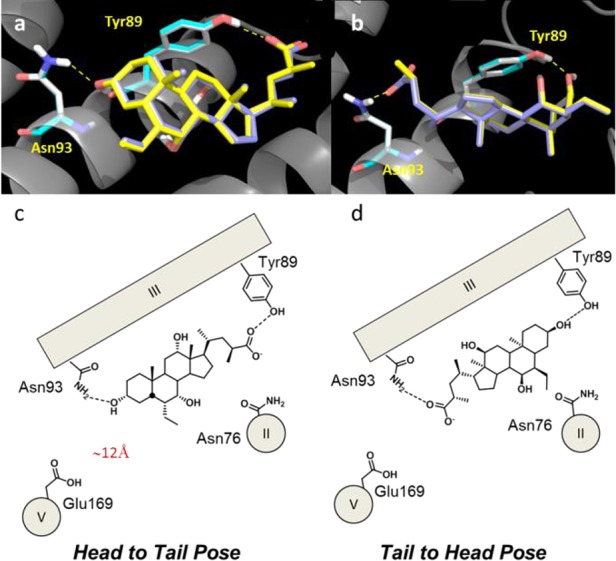
Three-dimensional views (a,b) and cartoons (c,d) showing the two working hypotheses of alternative binding poses obtained from docking experiments of 4 (yellow sticks in panels a and b) and 5 (blue sticks in panels a and b).
The biological profiling of BAs, including 4 and 5, confirmed the involvement of Ty89 in BAs binding (Table 1), whereas marginal effects were observed for TGR5Asn76Ala on the activity of BAs. Interestingly, on the basis of such results, it was still not possible to endorse one of the two binding pose possibilities. Indeed, BAs could bind to TGR5 with the carboxylic moiety making a hydrogen bond with Asn93 (binding mode 1, tail-to-head pose) or Tyr89 (binding mode 2, head-to-tail pose), and the hydroxyl group in position 3 interacting with Tyr89 or Asn93 (Figure 2). To address this issue, the novel bile amine 6 was designed, synthetized, and used as tool compound. The aim of 6 was to probe the head-to-tail pose, combining it with the engineering of additional mutant receptors: TGR5Asn93Asp, TGR5Tyr89Phe, and TGR5Tyr89Ala. If the head-to-tail pose was proven correct, then 6 would have shown an enhancement of the activity with respect to CDCA (2) in TGR5Tyr89Phe but not in TGR5Tyr89Ala, given the gaining of a cation−π interaction28 with Phe89 compensating the loss of the hydrogen-bonding interaction expected with the removal of the para-hydroxyl group of Tyr89 (Figure 3). Conversely, if the tail-to-head binding hypothesis was proven correct, then 6 would have shown an enhancement of the activity with respect to CDCA (2) in TGR5Asn93Asp, provided the engagement of an electrostatic interaction with Asp93.
Figure 3.
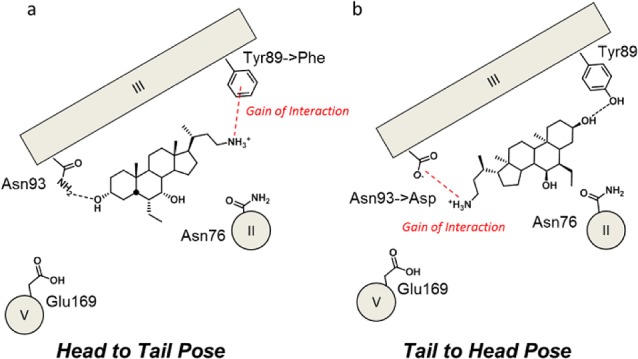
Cartoons showing the gain of function strategy with compound 6 to endorse a specific binding mode of BAs to TGR5.
As a first result, it was found that compound 6 is active at wild-type TGR5, showing a potency in the micromolar range (Table 1). Such an interesting result was also previously observed in the case of FXR, supporting the idea of an unconventional bioisosteric relationship between the amine group and the carboxylic moiety in bile acid receptors.29,30 While Asn93Asp mutation reduced the activity of 5, the activity was fully abolished in the case of compound 6, thereby disproving the tail-to-head binding pose. Conversely, supporting the head-to-tail hypothesis, Tyr89Phe mutation proved to keep the activity of compound 6 and 6-ECDCA (5), whereas Tyr89Ala disrupted the activity of both compounds (Table 1).
However, since phenylalanine lacks the para-hydroxyl group on the side chain, hydrophobic interactions were envisaged as occurring between Tyr89 and 5 as well as other BAs, which could not be fully explained on the basis of the proposed head-to-tail binding mode depicting hydrogen-bonding interactions with such residue (Figure 2).
Immuno-fluorescent (IF) staining experiments on CHO cells transfected with TGR5 mutants confirmed the above results, showing the expression of all mutant receptors as well as their localization on the membrane (see Supporting Information, Figure S3). Thus, on the basis of such results, the interaction model of BAs to TGR5 was further refined performing a cycle of energy minimization of binding site residues, followed by additional docking studies with a more extensive induced fit protocol on S-EMCA (4). Although five additional groups of binding poses of BAs to TGR5 were identified (binding modes 3–7; Table S3, Supporting Information), only one of them was compliant to mutagenesis data (binding mode 3; Figure S4a, Supporting Information) which also included the best scored pose (IFD = −634.1 kcal/mol). Indeed, the remaining binding modes either showed lack of hydrogen-bonding interaction with Asn93 (binding modes 5–7; Figure S4c, Supporting Information) or presence of hydrogen-bonding interaction with Asn76 (binding mode 4; Figure S4b, Supporting Information).
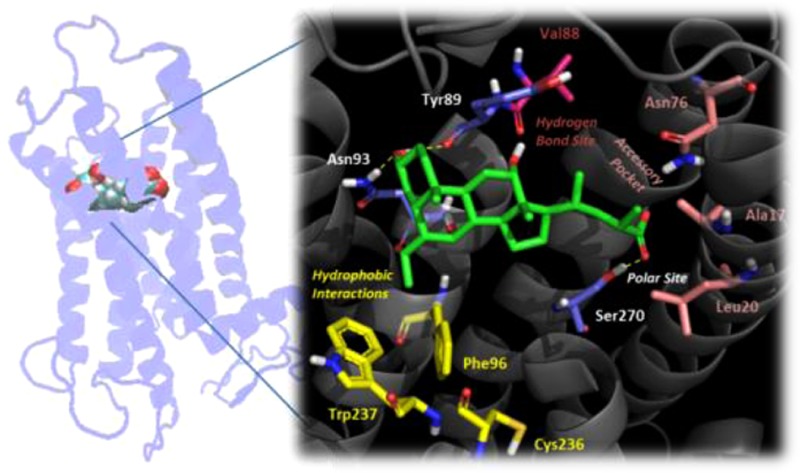
Hence, according to the head-to-tail binding mode 3 (Figure 4), the hydroxyl group in position C3 of 4 was involved in hydrogen-bonding interaction with the side chain of Asn93 and the backbone of Tyr89, while the aromatic side chain of Tyr89 was packing the steroid scaffold with hydrophobic interactions. The C6 ethyl group was lodged in a narrow hydrophobic pocket defined by the side chain of residues Phe96 and Trp237. This latter residue, in particular, is conserved in several GPCRs (W6.48) and reported to be a toggle switch for receptor activation.31 This would explain the high potency profile of C6 alkyl derivatives toward TGR5.19 Noteworthy, conversely from previous docking results (binding modes 1 and 2), S-EMCA (4) adopted an extended bioactive conformation rather than a folded one in the binding cleft of TGR5, with its carboxylic group making a hydrogen-bonding interaction with the side chain of Ser270. Of note, such residue is located close to Ser272 on helix TM7, a residue previously identified as a constitutive active variant (Ser272Gly) of TGR5 in primary sclerosing cholangitis (PSC) and proposed to be involved in mediating conformational changes of the receptor.23
Figure 4.
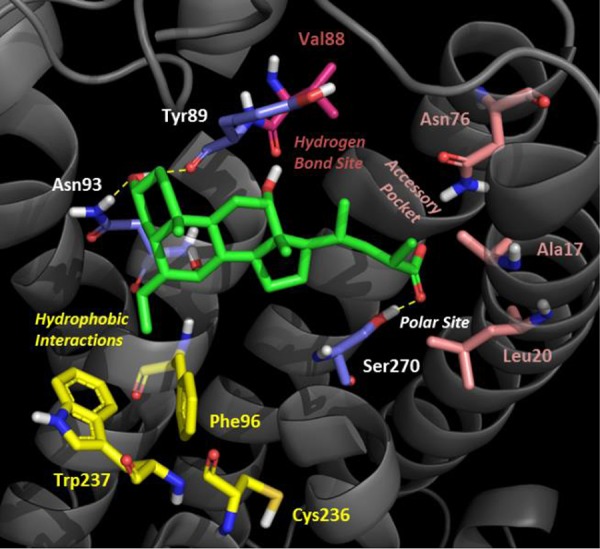
Binding mode of S-EMCA (4) to TGR5. Key residues involved in hydrogen-bonding interactions are color-coded in blue (Ty89, Asn93, and Ser270); residues lining the hydrophobic pocket are color-coded in yellow (Phe96, Cys236, and Trp237); residues defining the accessory pocket are color-coded in pink (Ala17, Leu20, and Asn76); the residue accounting for the potential hydrogen-bonding interaction site is color-coded in magenta (Val88).
To provide conclusive evidence to the study, a further TGR5Ser270Ala mutant was thus engineered and used to profile BAs. Biological profiling of BAs to this mutant receptor showed a complete loss of activity, eventually proving the binding pose of BAs to TGR5 according to a head-to-tail pose (Table 1). Of note, a fair agreement could be observed between the refined binding mode of BAs to TGR5 and our previous results of three-dimensional quantitative structure–activity relationship studies (3D QSAR) that evidenced four different regions around the BA scaffold as affecting TGR5 activity.20 In particular, the proposed large polar site recognizing the acidic side chain of BAs was identified in a large polar region of the receptor around Ser270. The hydrophobic pocket hosting the C6 and C7 positions of the BA steroid nucleus was defined by hydrophobic residues including Phe96, Cys236, and Trp237. According to our previous studies,20 this pocket accounts for potency of 4 and 5 over 2 and 3, respectively. Residues Ala17, Leu20, and Asn76 defined the accessory pocket lining the C-23(S) position of S-EMCA (4), which is responsible for the selectivity of 4 at TGR5 over FXR.14,15 Although a narrow hydrogen-bonding interaction site bridging the hydroxyl group in C12 of S-EMCA (4) was not observed, we envisaged the carbonyl of Val88 lining position C3 of the BA scaffold as potentially accounting for such a role.
In summary, we have described how BAs bind to TGR5 identifying key residues that mediate such interaction. In doing so, we have elucidated for the first time an experimentally validated complex of TGR5 with BAs that could be instrumental to generating new ideas for the development of novel potent ligands of the receptor.
Glossary
ABBREVIATIONS
- BAs
bile acids
- GPCRs
G protein-coupled receptors
- T2D
Type-2 diabetes
- WT
wild type
- TM
transmembrane helix
- EL
extracellular loop
- CA
cholic acid
- LCA
litocholic acid
- CDCA
chenodeoxycholic acid
- CHO
Chinese hamster ovary cells
- IFD
induced fit docking
- rmsd
root-mean-square deviation
- PSC
primary sclerosis cholangitis
Supporting Information Available
Experimental parts and results of docking experiments are reported in Tables S1–S3 and Figures S1, S2, and S4. Results of immunofluorescence staining experiments in CHO cells of TGR5 mutants and NMR spectra of 6 are reported in Figures S3 and S5, respectively. Figure S6 shows the energy minimization shells of the receptor. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by Intercept Pharmaceuticals (New York).
The authors declare no competing financial interest.
Supplementary Material
References
- Evans J. F.; Hutchinson J. H. Seeing the future of bioactive lipid drug targets. Nat. Chem. Biol. 2010, 67476–9. [DOI] [PubMed] [Google Scholar]
- Kawamata Y.; Fujii R.; Hosoya M.; Harada M.; Yoshida H.; Miwa M.; Fukusumi S.; Habata Y.; Itoh T.; Shintani Y.; Hinuma S.; Fujisawa Y.; Fujino M. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278119435–40. [DOI] [PubMed] [Google Scholar]
- Maruyama T.; Miyamoto Y.; Nakamura T.; Tamai Y.; Okada H.; Sugiyama E.; Itadani H.; Tanaka K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 2985714–9. [DOI] [PubMed] [Google Scholar]
- Thomas C.; Pellicciari R.; Pruzanski M.; Auwerx J.; Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug. Discovery 2008, 78678–93. [DOI] [PubMed] [Google Scholar]
- Pols T. W.; Noriega L. G.; Nomura M.; Auwerx J.; Schoonjans K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J. Hepatol. 2011, 5461263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C.; Auwerx J.; Schoonjans K. Bile acids and the membrane bile acid receptor TGR5: connecting nutrition and metabolism. Thyroid 2008, 182167–74. [DOI] [PubMed] [Google Scholar]
- Pols T. W.; Auwerx J.; Schoonjans K. Targeting the TGR5-GLP-1 pathway to combat type 2 diabetes and non-alcoholic fatty liver disease. Gastroenterol. Clin. Biol. 2010, 344–5270–3. [DOI] [PubMed] [Google Scholar]
- Pols T. W.; Nomura M.; Harach T.; Lo Sasso G.; Oosterveer M. H.; Thomas C.; Rizzo G.; Gioiello A.; Adorini L.; Pellicciari R.; Auwerx J.; Schoonjans K. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell. Metab. 2011, 146747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C.; Gioiello A.; Noriega L.; Strehle A.; Oury J.; Rizzo G.; Macchiarulo A.; Yamamoto H.; Mataki C.; Pruzanski M.; Pellicciari R.; Auwerx J.; Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell. Metab. 2009, 103167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prawitt J.; Caron S.; Staels B. Bile acid metabolism and the pathogenesis of type 2 diabetes. Curr. Diabetes Rep. 2011, 113160–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop F. K. Bile-induced secretion of glucagon-like peptide-1: pathophysiological implications in type 2 diabetes?. Am. J. Physiol. Endocrinol. Metab. 2010, 2991E10–3. [DOI] [PubMed] [Google Scholar]
- Lefebvre P.; Cariou B.; Lien F.; Kuipers F.; Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 891147–91. [DOI] [PubMed] [Google Scholar]
- Gioiello A.; Rosatelli E.; Nuti R.; Macchiarulo A.; Pellicciari R. Patented TGR5 modulators: a review (2006–present). Expert Opin. Ther. Pat. 2012, 22121399–414. [DOI] [PubMed] [Google Scholar]
- Pellicciari R.; Gioiello A.; Macchiarulo A.; Thomas C.; Rosatelli E.; Natalini B.; Sardella R.; Pruzanski M.; Roda A.; Pastorini E.; Schoonjans K.; Auwerx J. Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J. Med. Chem. 2009, 52247958–61. [DOI] [PubMed] [Google Scholar]
- Pellicciari R.; Sato H.; Gioiello A.; Costantino G.; Macchiarulo A.; Sadeghpour B. M.; Giorgi G.; Schoonjans K.; Auwerx J. Nongenomic actions of bile acids. Synthesis and preliminary characterization of 23- and 6,23-alkyl-substituted bile acid derivatives as selective modulators for the G-protein coupled receptor TGR5. J. Med. Chem. 2007, 50184265–8. [DOI] [PubMed] [Google Scholar]
- Herbert M. R.; Siegel D. L.; Staszewski L.; Cayanan C.; Banerjee U.; Dhamija S.; Anderson J.; Fan A.; Wang L.; Rix P.; Shiau A. K.; Rao T. S.; Noble S. A.; Heyman R. A.; Bischoff E.; Guha M.; Kabakibi A.; Pinkerton A. B. Synthesis and SAR of 2-aryl-3-aminomethylquinolines as agonists of the bile acid receptor TGR5. Bioorg. Med. Chem. Lett. 2010, 20195718–21. [DOI] [PubMed] [Google Scholar]
- Budzik B. W.; Evans K. A.; Wisnoski D. D.; Jin J.; Rivero R. A.; Szewczyk G. R.; Jayawickreme C.; Moncol D. L.; Yu H. Synthesis and structure–activity relationships of a series of 3-aryl-4-isoxazolecarboxamides as a new class of TGR5 agonists. Bioorg. Med. Chem. Lett. 2010, 2041363–7. [DOI] [PubMed] [Google Scholar]
- Evans K. A.; Budzik B. W.; Ross S. A.; Wisnoski D. D.; Jin J.; Rivero R. A.; Vimal M.; Szewczyk G. R.; Jayawickreme C.; Moncol D. L.; Rimele T. J.; Armour S. L.; Weaver S. P.; Griffin R. J.; Tadepalli S. M.; Jeune M. R.; Shearer T. W.; Chen Z. B.; Chen L.; Anderson D. L.; Becherer J. D.; De Los Frailes M.; Colilla F. J. Discovery of 3-aryl-4-isoxazolecarboxamides as TGR5 receptor agonists. J. Med. Chem. 2009, 52247962–5. [DOI] [PubMed] [Google Scholar]
- Sato H.; Macchiarulo A.; Thomas C.; Gioiello A.; Une M.; Hofmann A. F.; Saladin R.; Schoonjans K.; Pellicciari R.; Auwerx J. Novel potent and selective bile acid derivatives as TGR5 agonists: biological screening, structure–activity relationships, and molecular modeling studies. J. Med. Chem. 2008, 5161831–41. [DOI] [PubMed] [Google Scholar]
- Macchiarulo A.; Gioiello A.; Thomas C.; Massarotti A.; Nuti R.; Rosatelli E.; Sabbatini P.; Schoonjans K.; Auwerx J.; Pellicciari R. Molecular field analysis and 3D-quantitative structure-activity relationship study (MFA 3D-QSAR) unveil novel features of bile acid recognition at TGR5. J. Chem. Inf. Model. 2008, 4891792–801. [DOI] [PubMed] [Google Scholar]
- Genet C.; Schmidt C.; Strehle A.; Schoonjans K.; Auwerx J.; Saladin R.; Wagner A. Redefining the TGR5 triterpenoid binding pocket at the C-3 position. ChemMedChem 2010, 5121983–8. [DOI] [PubMed] [Google Scholar]
- Genet C.; Strehle A.; Schmidt C.; Boudjelal G.; Lobstein A.; Schoonjans K.; Souchet M.; Auwerx J.; Saladin R.; Wagner A. Structure–activity relationship study of betulinic acid, a novel and selective TGR5 agonist, and its synthetic derivatives: potential impact in diabetes. J. Med. Chem. 2010, 531178–90. [DOI] [PubMed] [Google Scholar]
- Hov J. R.; Keitel V.; Laerdahl J. K.; Spomer L.; Ellinghaus E.; ElSharawy A.; Melum E.; Boberg K. M.; Manke T.; Balschun T.; Schramm C.; Bergquist A.; Weismuller T.; Gotthardt D.; Rust C.; Henckaerts L.; Onnie C. M.; Weersma R. K.; Sterneck M.; Teufel A.; Runz H.; Stiehl A.; Ponsioen C. Y.; Wijmenga C.; Vatn M. H.; Stokkers P. C.; Vermeire S.; Mathew C. G.; Lie B. A.; Beuers U.; Manns M. P.; Schreiber S.; Schrumpf E.; Haussinger D.; Franke A.; Karlsen T. H. Mutational characterization of the bile acid receptor TGR5 in primary sclerosing cholangitis. PLoS One 2010, 58e12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R. E.; Bissantz C.; Gavelle O.; Kuratli C.; Dehmlow H.; Richter H. G.; Obst Sander U.; Erickson S. D.; Kim K.; Pietranico-Cole S. L.; Alvarez-Sanchez R.; Ullmer C. 2-Phenoxy-nicotinamides are potent agonists at the bile acid receptor GPBAR1 (TGR5). ChemMedChem 2013, 84569–76. [DOI] [PubMed] [Google Scholar]
- Pellicciari R.; Gioiello A.; Sabbatini P.; Venturoni F.; Nuti R.; Colliva C.; Rizzo G.; Adorini L.; Pruzanski M.; Roda A.; Macchiarulo A. Avicholic acid: A lead compound from birds on the route to potent TGR5 modulators. ACS Med. Chem. Lett. 2012, 34273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hov J. R.; Keitel V.; Schrumpf E.; Haussinger D.; Karlsen T. H. TGR5 sequence variation in primary sclerosing cholangitis. Dig. Dis. 2011, 29178–84. [DOI] [PubMed] [Google Scholar]
- Surgand J. S.; Rodrigo J.; Kellenberger E.; Rognan D. A chemogenomic analysis of the transmembrane binding cavity of human G-protein-coupled receptors. Proteins 2006, 622509–38. [DOI] [PubMed] [Google Scholar]
- Deakyne C. A.; Meot-Ner M. Unconventional ionic hydrogen bonds. 1. CHδ+···X. Complexes of quaternary ions with n- and π-donors. J. Am. Chem. Soc. 1985, 1072469–474. [Google Scholar]
- Macchiarulo A.; Nuti R.; Eren G.; Pellicciari R. Charting the chemical space of target sites: insights into the binding modes of amine and amidine groups. J. Chem. Inf. Model. 2009, 494900–12. [DOI] [PubMed] [Google Scholar]
- Macchiarulo A.; Pellicciari R. Exploring the other side of biologically relevant chemical space: insights into carboxylic, sulfonic and phosphonic acid bioisosteric relationships. J. Mol. Graphics Model. 2007, 264728–39. [DOI] [PubMed] [Google Scholar]
- Trzaskowski B.; Latek D.; Yuan S.; Ghoshdastider U.; Debinski A.; Filipek S. Action of molecular switches in GPCRs: theoretical and experimental studies. Curr. Med. Chem. 2012, 1981090–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.