

Abstract

Hyperactive signaling of the MAP kinase pathway resulting from the constitutively active B-RafV600E mutated enzyme has been observed in a number of human tumors, including melanomas. Herein we report the discovery and biological evaluation of GSK2118436, a selective inhibitor of Raf kinases with potent in vitro activity in oncogenic B-Raf-driven melanoma and colorectal carcinoma cells and robust in vivo antitumor and pharmacodynamic activity in mouse models of B-RafV600E human melanoma. GSK2118436 was identified as a development candidate, and early clinical results have shown significant activity in patients with B-Raf mutant melanoma.
Keywords: B-Raf, GSK2118436, dabrafenib, melanoma, MAP kinase
The MAP kinase pathway plays a central role in cellular growth processes, and aberrant signaling through this pathway has been observed in many cancers.1−88 Notably, activating mutations in B-Raf have been identified in more than 60% of all melanomas.1 The most common mutation, observed in 80–90% of B-Raf mutant cancers, is the B-RafV600E mutation, which renders the kinase constitutively active. This mutation, which substitutes valine with glutamic acid at amino acid 600 in the activation loop, mimics regulatory phosphorylation and results in in vitro kinase activity of the protein being 500-fold greater than that of wild-type B-Raf.1,3 Because tumors bearing an activating B-RafV600E mutation show oncogenic addiction to this hyperactivated signaling pathway, it was postulated that small molecule inhibitors of B-RafV600E kinase activity offer a novel, targeted approach for the treatment of B-RafV600E-driven cancers.4 This hypothesis has since been tested clinically, and significant responses to selective B-Raf inhibitor therapy have been observed.5
Herein we describe the medicinal chemistry efforts leading to the discovery of GSK2118436 (dabrafenib), a potent, selective, and efficacious inhibitor of B-RafV600E. GSK2118436 is currently in phase III clinical development for the treatment of activating B-Raf mutant tumors.
Our early efforts aimed at discovering inhibitors of B-RafV600E suitable for clinical evaluation led to the identification of thiazole 1.6 This compound shows potent in vitro kinase inhibitory activity against the B-RafV600E enzyme, and in both cellular mechanistic (pERK) and proliferation assays in the B-RafV600E SKMEL28 melanoma cell line (Table 1). Thiazole 1 also had low clearance and good overall oral systemic exposure in rats. However, this compound displayed very high clearance and poor bioavailability in nonrodent species. Additionally, metabolite identification studies conducted in dog and monkey liver microsomes identified several major metabolites clustered in the tail and core regions of the molecule. We hypothesized that drug exposure after an oral dose in the higher species was limited by rapid compound metabolism. Medicinal chemistry efforts were thus focused on improving pharmacokinetic properties in dog while preserving the favorable target potency, selectivity, and rodent pharmacokinetic properties inherent in this template.7
Table 1. Structure, in Vitro Data, and Pharmacokinetic Data for Compound 1 in Rat, Dog, and Monkey.
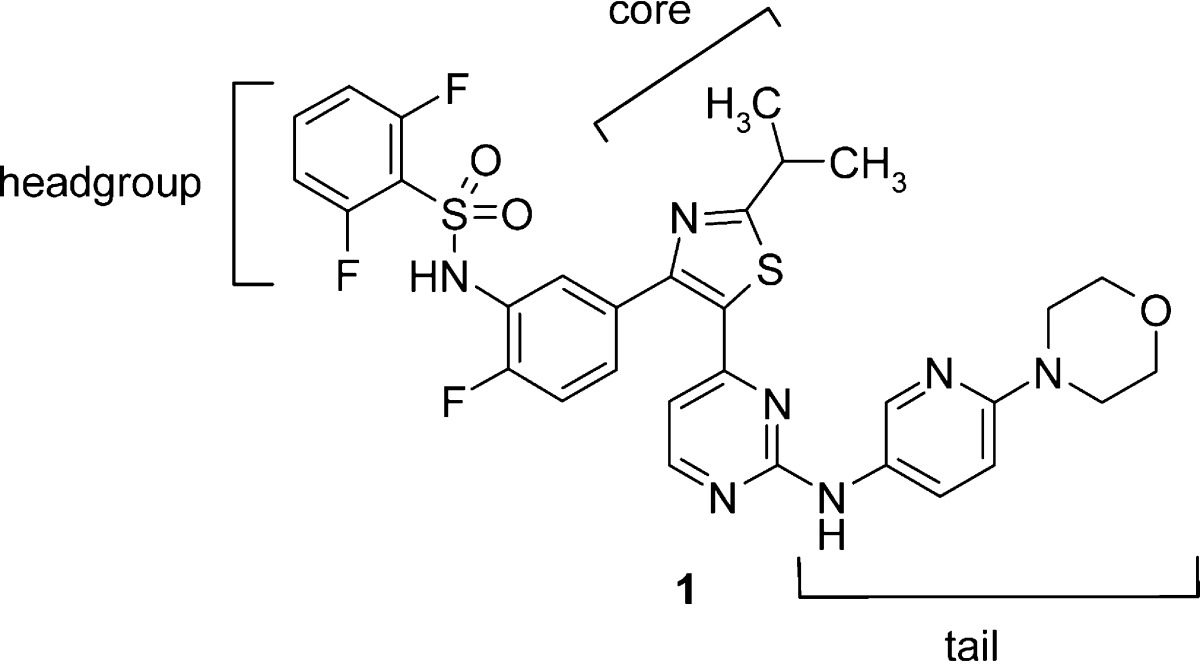
| B-RafV600E IC50 (nM) | pERK IC50 (nM) | SKMEL28 proliferation IC50 (nM) | species | intrinsic Clia | Clb | po DNAUCc | % F |
|---|---|---|---|---|---|---|---|
| 3.8 | 23 | 48 | rat | 9 | 9d | 1748d | 85 |
| dog | 53 | 38e | 42e | 10 | |||
| monkey | 8 | 39e | 23e | 5 |
Intrinsic clearance data from liver microsomes, reported in units of milliliters per minute per gram of liver.
Intravenous blood clearance reported in units of milliliters per minute per kilogram.
Oral dose-normalized AUC (DNAUC) reported as nanograms hour per milliliter per milligram per kilogram (n = 3).
Dosing vehicle: 1% DMSO, 10% solutol in saline or water, at pH 5.0.
Dosing vehicle: 1% DMSO, 30% PEG400 in saline or water, at pH 3.5–4.5.
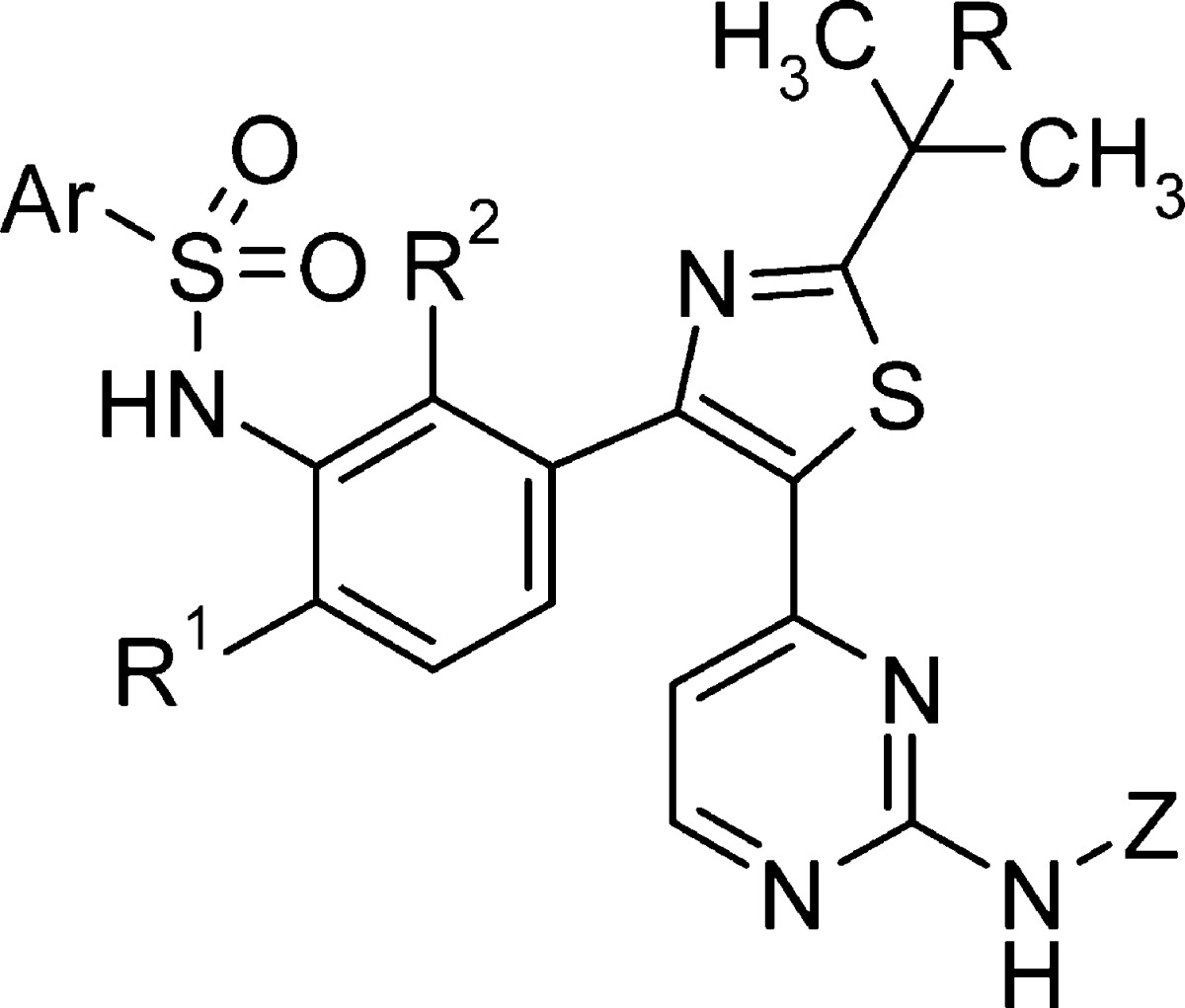
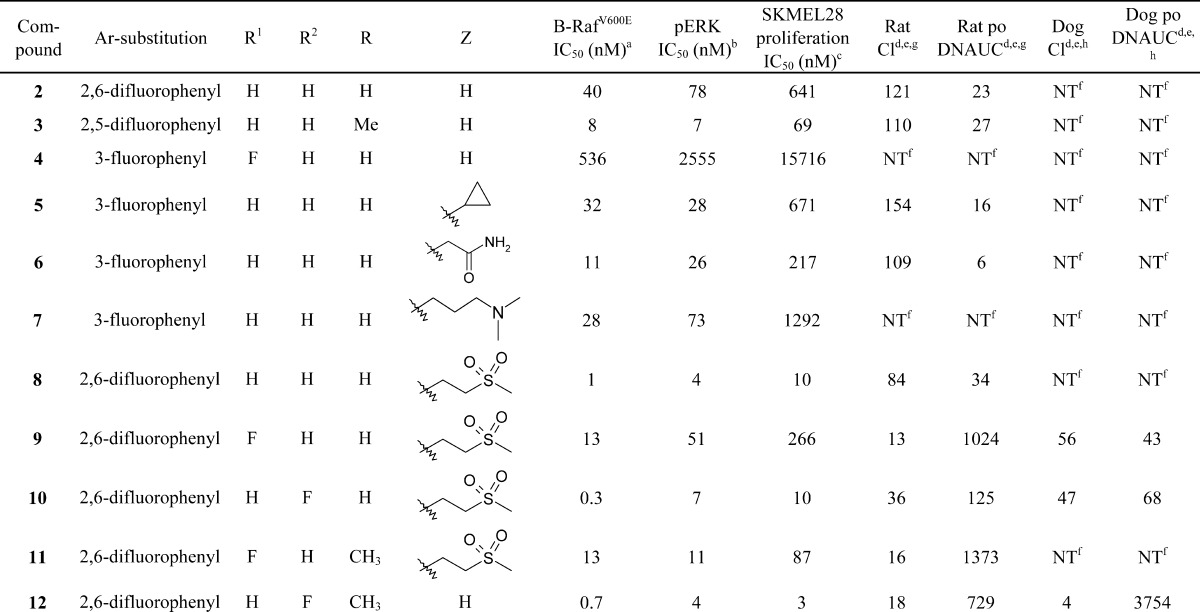
Having established the sulfonamide as a key pharmacophore required for potent cellular inhibition of B-RafV600E, we performed significant structural modifications elsewhere to lower the molecular weight and reduce the number of metabolic sites contained within the template. Truncation of the tail by replacing the aminopyridine in 1 with a small alkyl group (5) or polar functionality (6–8) largely preserved enzyme potency, cellular mechanistic potency (pERK), and inhibition of proliferation in B-RafV600E SKMEL28 cells but resulted in significantly higher clearance in rat (Table 2).8 Surprisingly, unsubstituted aminopyrimidine hinge binder (2) maintained in vitro potency against B-Raf;9 however, this compound also demonstrated high in vivo clearance in rat. Core group modifications were attempted in an effort to alter the metabolic stability of this series, leading to a tert-butyl core analogue (3). While this change from isopropyl to tert-butyl resulted in a noteworthy increase in potency, rat intravenous (iv) clearance was still very high and oral exposure was commensurately low. Since fluoro substitution at position 6 of the phenyl ring [R1 = F (Table 2)] led to an important improvement in rat pharmacokinetic properties in previous SAR efforts,6 this substitution was attempted in the truncated tail series (4). Unfortunately, in this context, fluoro substitution at R1, when combined with the tail truncation, led to a decrease in potency such that further progression of compound 4 was abandoned.
Table 2. Truncated Tail SAR.
B-RafV600E enzymatic activity. Values are means of at least two experiments; individual values are within 2-fold of the reported mean value.
Inhibition of pERK in SKMEL28 cells. Values are means of at least two experiments; individual values are within 2-fold of the reported mean value.
Inhibition of SKMEL28 cell proliferation. IC50 values are means of at least experiments; individual values are within 3-fold of the reported mean value.
Intravenous clearance reported in units of milliliters per minute per kilogram. Oral dose-normalized AUC (DNAUC) reported as nanograms hours per milliliter per milligram per kilogram (n = 3).
Data from four-compound cassette dosing. Pharmacokinetic values based on cassette dosing were found to be within 2-fold of those of discretely dosed experiments.
Not tested.
Dosing vehicle: 1% DMSO, 10% solutol in saline or water, at pH 5.0.
Dosing vehicle: 1% DMSO, 30% PEG400 in saline or water, at pH 3.5–4.5.
Given the favorable potency demonstrated with compound 8, SAR in this series was further explored. In an effort to lower IV clearance in rats, compounds with substitution at positions R, R1, and R2 were prepared and evaluated. Fluorination at R1 (9) decreased the enzyme and cellular potency but resulted in a marked improvement in rat iv clearance and oral exposure. Further SAR on this template showed that fluorination at R2 (10) improved or maintained the enzyme and cellular potency and resulted in improved pharmacokinetic properties compared to those of the des-fluoro analogue (8). Replacement of the isopropyl thiazole core with a tert-butyl thiazole core (11) also resulted in improved cellular potency. While rat pharmacokinetic properties could be improved by extrapolating previous SAR findings from this series, analogous improvements in dog pharmacokinetic properties were not realized, as compounds 9 and 10 both displayed high clearance and poor oral exposure in dog.
Further attempts to optimize pharmacokinetic properties while maintaining potency in this series were made by combining the structural modifications of fluorination at R2, replacing the isopropyl core with a tert-butyl group, and utilizing the unsubstituted aminopyrimidine tail group from compounds 2–4. Through this effort, compound 12 (GSK2118436) was synthesized and evaluated (Table 2 and Figure 1).
Figure 1.

Dabrafenib (GSK2118436).
GSK2118436 (12) displayed compelling inhibitory activity in enzyme and cellular mechanistic assays, and in cell proliferation assays in B-RafV600E-driven melanoma lines, SKMEL28 and A375P F11 (IC50 = 3 and 8 nM, respectively), and colorectal carcinoma line Colo205 (IC50 = 7 nM). Gratifyingly, GSK2118436 was found to have a minimal effect in vitro on cells with wild-type B-Raf (HFF IC50 = 3.0 μM) and in tumor cells not harboring the activating B-RafV600E mutation.10 Additionally, acceptable rat and dramatically improved dog pharmacokinetic profiles were observed, in contrast to those of previously prepared analogues.
The remarkable improvement in dog pharmacokinetic properties prompted further investigation in order to understand the rationale for the finding. In an effort to understand the high clearance observed in dog from sulfone-containing tail compounds (Table 2), 9 was incubated in dog hepatocytes and the major metabolites were identified by mass spectrometry (Figure 2). Following incubation of compound 9 in dog hepatocytes, two major metabolites were identified: compound 13, showing complete dealkylation of the ethylmethylsulfone tail, and compound 14, a product of oxidation of the isopropyl core. In retrospect, it was surmised that removal of the tail group and replacement of the isopropyl core group with a tert-butyl attenuated the metabolic liabilities associated with these positions and led to the significant improvements in the dog pharmacokinetic properties of 12.
Figure 2.

Metabolite identification of 9 in dog hepatocytes.
GSK2118436 was screened against a set of 61 kinases representing broad coverage of the kinome and was found to be a potent biochemical inhibitor of B-RafV600E, wild-type B-Raf, and c-Raf, displaying subnanomolar or nanomolar potencies (see the Supporting Information). Importantly, GSK2118436 was also found to be highly selective, exhibiting >500-fold selectivity for B-RafV600E compared to most kinases screened.11 Significant activity (<100-fold selectivity) was observed for a single kinase in the panel, Alk5. The impact of Alk5 enzyme activity on cellular signaling was further investigated by measuring the downstream phosphorylation level of SMAD2/3 in HepG2 cells.12 GSK2118436 was significantly less effective at inhibiting SMAD2/3 phosphorylation (IC50 = 3.7 μM) compared with inhibiting ERK phosphorylation (IC50 = 4 nM) in a cellular context. These data underscore the remarkable selectivity achieved by this uniquely potent and selective inhibitor of B-RafV600E.
GSK2118436 was also studied in vivo and found to dramatically reduce tumor growth in mice bearing B-RafV600E human melanoma tumors. In this model, CD1 nu/nu mice bearing A375P F11 (B-RafV600E) tumors were dosed orally with GSK2118436 at doses of 0.1, 1, 10, and 100 mg/kg once daily for 14 days (Figure 3). Dose-proportional reductions in tumor growth were observed. Body weight was also measured, and no significant changes were observed at any of the doses tested. Notably, in the 100 mg/kg group, complete tumor regression was observed in 50% of treated animals.
Figure 3.

In vivo efficacy of GSK2118436 in CD1 nu/nu mice bearing A375P F11 (B-RafV600E) tumors (n = 8 per group).
Additionally, GSK2118436 reduced the levels of pERK in A375P F11 (B-RafV600E) tumor tissue in vivo in a dose-dependent manner after a single oral dose. Tumors were collected 2, 6, and 24 h postdose, and pERK levels in each were measured and normalized to the total ERK (tERK) present. The MEK inhibitor PD0325901 was used as a control. Figure 4 shows pERK/tERK levels compared to those of vehicle-treated animals and pharmacokinetic data in a composite of two separate studies. Levels of pERK/tERK were substantially reduced 2 and 6 h postdose, with a notable pharmacodynamic effect (>50% inhibition) at doses of ≥3 mg/kg in this single-dose study, which returned to untreated levels by 24 h at doses of ≤30 mg/kg. The concentration of drug measured in the blood and tumor during the course of the study correlated with the observed pharmacodynamic effects. These data suggest that under the conditions tested, a single oral dose of ≥3 mg/kg GSK2118436 can maintain ≥50% B-Raf inhibition for at least 6 h.
Figure 4.

In vivo pharmacodynamic response of a single dose of GSK2118436 in CD1 nu/nu mice bearing A375P F11 (B-RafV600E) tumors (n = 4 per group).
In summary, lead molecule 1, with high potency and poor pharmacokinetic properties in higher species, was rapidly evolved into clinical development candidate GSK2118436 (12). Identification of the key B-RafV600E potency contributors (sulfonamide head, R2 fluorination, and tert-butyl thiazole core) through detailed and iterative SAR studies allowed for the elimination of nonessential portions of the lead structure and high selectivity to be achieved across the kinome. With high enzyme and cellular potency engineered into the original lead, pharmacokinetic issues were addressed by combining multiple changes in the template, resulting in the improved multispecies PK properties observed for 12. With high rodent exposure realized in GSK2118436, we were able to demonstrate target-related pharmacology in two in vivo models, gaining a high level of confidence in the progression of this molecule into clinical studies. Additionally, vast improvements in higher-species PK allowed for the achievement of exposures necessary to fully explore the safety of the molecule preclinically. Finally, a >20% decrease in molecular weight compared to that of 1 resulted in improved physicochemical properties, expediting the identification of a salt version (mesylate) and form suitable for early clinical studies.
The efforts described herein ultimately led to the discovery of GSK2118436 (dabrafenib), which is currently in advanced clinical studies in B-Raf mutant tumors. GSK2118436 has shown remarkable efficacy in melanoma patients with activating B-Raf mutations, including patients with brain metastases, and has further shown enhanced clinical activity in combination with the MEK inhibitor, trametinib.13−33 Details of these studies will be reported in due course.
Acknowledgments
The authors thank Erich Baum, Neil Bifulco, Ronda Davis-Ward, Kelly Donaldson, Kyle Emmitte, Phil Harris, Kevin Kuntz, Kristen Nailor, Jim Salovich, Doug Sammond, Greg Schaaf, Stephon Smith, and Brian Wilson for scientific support and Dash Dhanak and Dirk Heerding for critically reviewing the manuscript and support throughout the duration of the program.
Supporting Information Available
Biochemical and cellular assays, metabolite identification experimental procedures, kinase selectivity data, A375P F11 melanoma xenograft, PK analysis, pharmacodynamic measurement of pERK in tissues, and experimental procedures for the compounds and characterization of dabrafenib. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interests: T.R.R., S.H.D., K.G.P., S.G.L., M.R.A., K.N.S., L.S.K.-C., and K.G.M. are current employees and stockholders of GlaxoSmithKline.
Notes
All studies involving the use of animals were conducted after review by the GlaxoSmithKline (GSK) Institutional Animal Care and Use Committee and in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals.
Supplementary Material
References
- Davies H.; Bignell G.; Cox C.; Stevens P.; Edkins S.; Clegg S.; Teague J.; Woffendin H.; Garnett M.; Bottomley W.; Davis N.; Dicks E.; Ewing R.; Floyd Y.; Gray K.; Hall S.; Hawes R.; Hughes J.; Kosmidou V.; Menzies A.; Mould C.; Parker A.; Stevens C.; Watt S.; Hooper S.; Wilson R.; Jayatilake H.; Gusterson B.; Cooper C.; Shipley J.; Hargrave D.; Pritchard-Jones K.; Maitland N.; Chenevix-Trench G.; Riggins G.; Bigner D.; Palmieri G.; Cossu A.; Flanagan A.; Nicholson A.; Ho J.; Leung S.; Yuen S.; Weber B.; Seigler H.; Darrow T.; Paterson H.; Marais R.; Marshall C.; Wooster R.; Stratton M.; Futreal P. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [DOI] [PubMed] [Google Scholar]
- Singer G.; Oldt R.; Cohen Y.; Wang B.; Sidransky D.; Kurman R.; Shih Ie. M. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [DOI] [PubMed] [Google Scholar]
- Cohen Y.; Xing M.; Mambo E.; Guo Z.; Wu G.; Trink B.; Beller U.; Westra W. H.; Ladenson P. W.; Sidransky D. BRAF mutation in papillary thyroid carcinoma. J. Natl. Cancer Inst. 2003, 95, 625–627. [DOI] [PubMed] [Google Scholar]
- Yuen S.; Davies H.; Chan T.; Ho J.; Bignell G.; Cox C.; Stephens P.; Edkins S.; Tsui W.; Chan A.; Futreal P.; Stratton M.; Wooster R.; Leung S. Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res. 2002, 62, 6451–6455. [PubMed] [Google Scholar]
- Wan P.; Garnett M.; Roe S.; Lee S.; Niculescu-Duvaz D.; Good V.; Jones C. M.; Marshall C. J.; Springer C. J.; Barford D.; Marais R. Mechanism of Action of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [DOI] [PubMed] [Google Scholar]
- Tsai J.; Lee J.; Wang W.; Zhang J.; Cho H.; Mamo S.; Bremer R.; Gillette S.; Kong J.; Haass N.; Sproesser K.; Li L.; Smalley K.; Fong D.; Zhu Y.-L.; Marimuthu A.; Nguyen H.; Lam W.; Liu J.; Cheung I.; Rice J.; Suzuki Y.; Luu C.; Settachatgul C.; Shellooe R.; Cantwell J.; Kim S.-H.; Schlessinger J.; Zhang K.; West B.; Powell B.; Habets G.; Zhang C.; Ibrahim P.; Hirth P.; Artis D.; Herlyn M.; Bollag G. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman P.; Hauschild A.; Robert C.; Haanen J.; Ascierto P.; Larkin J.; Dummer R.; Garbe C.; Testori A.; Maio M.; Hogg D.; Lorigan P.; Lebbe C.; Jouary T.; Schadendorf D.; Ribas A.; O’Day S.; Sosman J.; Kirkwood J.; Eggermont A.; Dreno B.; Nolop K.; Li J.; Nelson B.; Hou J.; Lee R.; Flaherty K. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen J.; Adjabeng G.; Arnone M.; Dickerson S.; Han C.; Hornberger K.; King A.; Mook R.; Petrov K.; Rheault T.; Rominger C.; Rossanese O.; Smitheman K.; Waterson A.; Uehling D. Development of Potent B-RafV600E Inhibitors Containing an Arylsulfonamide Headgroup. Bioorg. Med. Chem. Lett. 2011, 21, 4436–4440. [DOI] [PubMed] [Google Scholar]
- Efforts were focused on optimizing pharmacokinetic properties in rat and dog to identify two species for preclinical safety studies.
- Previously, we reported that fluorination of the arylsulfonamide headgroup at the 2-, 3-, 2,5-, and 2,6-positions gave optimal, and nearly interchangeable, potency in this series. Additionally, in vivo pharmacokinetic studies in rat and dog with similarly paired analogues showed that changes in the headgroup substitution pattern had little impact on either clearance or oral absorption. Thus, potency and pharmacokinetic properties observed in this series do not appear to be sensitive to changes in the fluorination pattern of the headgroup. See ref (6).
- For a discussion of the binding efficiency of truncated tail-containing molecule 2 compared to 1 in the context of the B-Raf binding mode, see the Supporting Information.
- King A., ; Arnone M., ; Bleam M., ; Moss K., ; Yang J., ; Fisher K., ; Smitheman K., ; Erhardt J.; Hughes-Earle A., ; Kane-Carson L., ; Sinnamon R., ; Qi H., ; Rheault T., ; Uehling D., ; Laquerre S., . For a thorough biological characterization of dabrafenib, see: Novel Selective BRAF Inhibitor (GSK2118436; dabrafenib) with Demonstration of Increased Efficacy and Reduced Skin Lesions by Combining BRAF and MEK Inhibitors. Manuscript in preparation.
- Broader kinase profiling was recently conducted on a set of 270 kinases (Millipore), and <100-fold activity was observed for only LIMK1, NEK11, PKD2, and SIK in addition to ALK5. See ref (10).
- Gellibert F.; de Gouville A.-C.; Woolven J.; Mathews N.; Nguyen V.-L.; Bertho-Ruault C.; Patikis A.; Grygielko E.; Laping N.; Huet S. Discovery of 4-{4-[3-(Pyridin-2-yl)-1H-pyrazol-4-yl]pyridin-2-yl}-N-(tetrahydro-2H-pyran-4-yl)benzamide (GW788388): A Potent, Selective, and Orally Active Transforming Growth Factor-β Type I Receptor Inhibitor. J. Med. Chem. 2006, 49, 2210–2221. [DOI] [PubMed] [Google Scholar]
- Hauschild A.; Grob J.; Demidov L.; Jouary T.; Gutzmer R.; Millward M.; Rutkowski P.; Blank C.; Miller W.; Kaempgen E.; Martín-Algarra S.; Karaszewska B.; Mauch C.; Chiarion-Sileni V.; Martin A.; Swann S.; Haney P.; Mirakhur B.; Guckert M.; Goodman V.; Chapman P. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [DOI] [PubMed] [Google Scholar]
- Falchook G.; Long G.; Kurzrock R.; Kim K.; Arkenau T.; Brown M.; Hamid O.; Infante J.; Millward M.; Pavlick A.; O’Day S.; Blackman S.; Curtis C.; Lebowitz P.; Ma B.; Ouellet D.; Kefford R. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 2012, 379, 1893–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J.; Flaherty K.; Infante J.; Falchook G.; Kefford R.; Daud A.; Hamid O.; Gonzalez R.; Kudchadkar R.; Lawrence D.; Burris H.; Long G.; Algazi A.; Lewis K.; Kim K.; Puzanov I.; Sun P.; Little S.; Patel K.; Sosman J. Updated safety and efficacy results from a phase I/II study of the oral BRAF inhibitor dabrafenib (GSK2118436) combined with the oral MEK 1/2 inhibitor trametinib (GSK1120212) in patients with BRAFi-naive metastatic melanoma. ASCO Meeting Abstracts 2012, 30, 8510. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.