

Abstract

PI3K, AKT, and mTOR are key kinases from PI3K signaling pathway being extensively pursued to treat a variety of cancers in oncology. To search for a structurally differentiated back-up candidate to PF-04691502, which is currently in phase I/II clinical trials for treating solid tumors, a lead optimization effort was carried out with a tricyclic imidazo[1,5]naphthyridine series. Integration of structure-based drug design and physical properties-based optimization yielded a potent and selective PI3K/mTOR dual kinase inhibitor PF-04979064. This manuscript discusses the lead optimization for the tricyclic series, which both improved the in vitro potency and addressed a number of ADMET issues including high metabolic clearance mediated by both P450 and aldehyde oxidase (AO), poor permeability, and poor solubility. An empirical scaling tool was developed to predict human clearance from in vitro human liver S9 assay data for tricyclic derivatives that were AO substrates.
Keywords: PF-04979064, kinase inhibitor, PI3K/mTOR dual inhibitor, aldehyde oxidase metabolism, cancer, antitumor
The phosphatidylinositol 3-kinase (PI3K) signaling pathway plays crucial roles in cell growth, proliferation, and survival and is a frequently dysregulated pathway in human cancers.1,2 Inhibitors of key kinases in the pathway, including PI3K, AKT, and mTOR, have been extensively pursued in oncology in recent years.3 Because PI3K/mTOR dual inhibitors may most effectively block the PI3K pathway, overcome feedback loops,4 and block PI3K-independent mTOR activation, our strategy to target PI3K signaling pathway was focused on PI3K/mTOR dual inhibitors. Two highly potent and selective ATP competitive kinase inhibitors of class 1 PI3Ks and mTOR from Pfizer, an oral agent PF-046915025,6 and an iv-administered agent PF-05212384 (PKI-587),7 are currently in phase I/II clinical trials for the treatment of solid tumors (Figure 1).
Figure 1.

Chemical structures of PF-04691502, PF-05212384, and the new lead 1.
PF-04691502 is derived from the 4-methylpyridopyrimidinone series. In our search for a structurally differentiated back-up candidate to PF-04691502, compound 1, from a tricyclic imidazo[1,5]naphthyridine series8,8b that was designed through a fast follower approach to BEZ235,9 was identified as an interesting lead. It exhibited potent in vitro activity against mouse PI3Kα,5 which was used as a surrogate of human PI3Kα in the primary screening, with a Ki of 1.41 nM, which translates to a ligand efficiency (LE) of 0.354. When tested in an mTOR kinase domain in vitro biochemical assay, 1 also exhibited good activity with a Ki of 4.51 nM. In a BT20 cell assay, measuring inhibition of AKT phosphorylation at S473, 1 exhibited moderate cellular potency with an IC50 of 144 nM.
Shown in Scheme 1 is the general synthetic route for preparing the tricyclic derivatives discussed in this paper.10 Readily available starting material 2 is reacted with 2-(ethoxymethylene)-malonate at elevated temperature to afford compound 3, which is heated in phenyl ether (Ph2O) to afford the cyclized compound 4. Treatment of 4 with POCl3 yields 4,6-dichloro-(1,5)-naphthyridine-3-carboxylic acid ethyl ester 5, which is subjected to Suzuki coupling conditions with a boronic acid or boronic ester derivative to give compound 6. Compound 7 is prepared by treatment of 6 with an amine, and subsequent hydrolysis of the ester in 7 gives the free acid 8. When 8 is treated with diphenylphosphoryl azide (DPPA) and Et3N, the initially formed nitrene intermediate from Curtius rearrangement reacts with the amine at 4-naphthyridine to undergo intramolecular cyclization to produce compound 9, which is methylated to give compound 10. The tricyclic derivatives with acylated piperidine moieties can be prepared from 10 by first removing the protecting groups on the piperidine, followed by amide formation.
Scheme 1. General Synthetic Route for the Tricyclic Derivatives.

Reagents: Step 1: 2-(ethoxymethylene)-malonate, ethanol, reflux. Step 2: phenyl ether, reflux. Step 3: POCl3, reflux. Step 4: boronic acid/boronic ester, K2CO3, Pd(PPh3)4, toluene. Step 5: R1NH2, AcOH. Step 6: LiOH, EtOH–water. Step 7: DPPA, Et3N, DMF. Step 8: MeI, NaOH, CH2Cl2.
Docking studies of the tricyclic analogues with PI3Kα were carried out,9 and the proposed binding mode for 1 is illustrated in Figure 2. The naphthyridine ring nitrogen formed a key hydrogen bond with the hinge residue Val 882. The ring nitrogen on the methoxypyridine formed a hydrogen bond with the conserved water molecule in the selectivity pocket. The phenyl group from the benzyl nitrile moiety bound in a hydrophobic pocket, and the nitrile formed a H bond with Lys 833 in the phosphate binding pocket.
Figure 2.
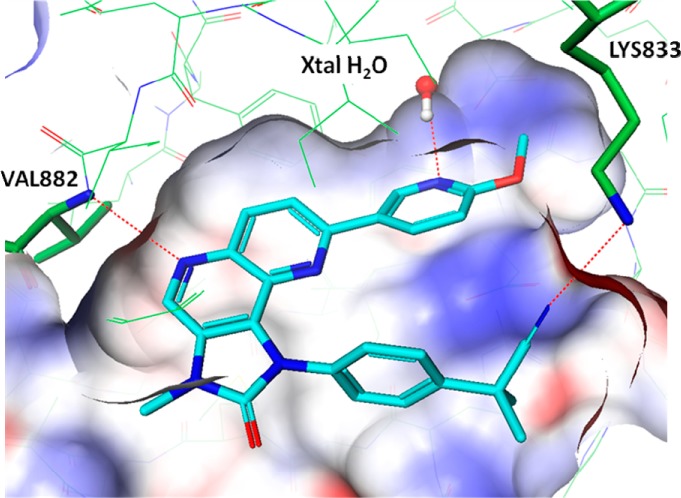
Modeled structure of compound 1 with PI3Kγ.
Physical properties-based optimization (PPBO) of multiple druglike attributes,11 guided by structure-based drug design (SBDD), commenced. Compound 1 was highly lipophilic, with a clogP of 4.69, exhibiting high metabolic clearance in both HLM and RLM in vitro assays, with extraction ratios (ERs) of 0.652 and 0.724, respectively. Compound 1 demonstrated poor solubility and poor permeability, with measured kinetic solubility of 2.00 μM5 and measured RRCK permeability of 0.305 × 10–6 cm/s, respectively. Even though 1 exhibited potent biochemical enzyme Ki against both mPI3Kα and mTOR, it only showed moderate cell potency; poor solubility and permeability of 1 may account for the high ratio between cell IC50 and mPI3Kα Ki values.
The medicinal chemistry goals for the lead optimization from 1 were to increase cell potency, metabolic stability, permeability, and solubility. Because compounds with lower clogP usually exhibit lower metabolic clearance and greater solubility,5 the strategy to increase metabolic stability and solubility was to reduce clogP for the new designs. In addition, 1 had multiple aromatic rings, and π–π stacking of the aromatic rings could also contribute to its poor solubility. Consequently, a second strategy to increase solubility was to increase the 3D structure to minimize π–π stacking. It was reasoned that increased solubility could also lead to improved permeability, which could help reduce the ratio between cell IC50 and enzymatic Ki. On the basis of the docking studies, because the phenyl group off the tricyclic core in 1 bound in a hydrophobic pocket, replacing the phenyl ring with a piperidine or a cyclohexane group would both maintain the hydrophobic interactions between the methylene groups of the piperidine or cyclohaxane ring with the enzyme and increase the 3D structure. In addition, polar groups such as an amide or an alcohol could also be introduced to the piperidine or cyclohexane ring to reduce clogP and still maintain the binding affinity by forming H bonds with the polar residues in the phosphate binding pocket.
Following the above-discussed strategies and tactics, tricyclic derivatives 11–14 in Table 1 were designed by replacing the benzyl nitrile moiety with substituted cyclohexane and piperidine derivatives. The clogP for these compounds ranged from 1.07 to 2.37. Data for 1 are shown in Table 1 for comparison. Overall, these compounds exhibited increased permeability and cellular potency. Significant increases in lipophilic efficiency (LipE = pIC50 – clogP)5 were achieved because clogP was significantly reduced by replacing the benzyl nitrile moiety with cyclohexane and piperidine derivatives. Metabolic clearance for compounds 12–14 was also reduced; however, solubility for 12 and 14 needed further improvement.
Table 1. Tricyclic Derivatives.
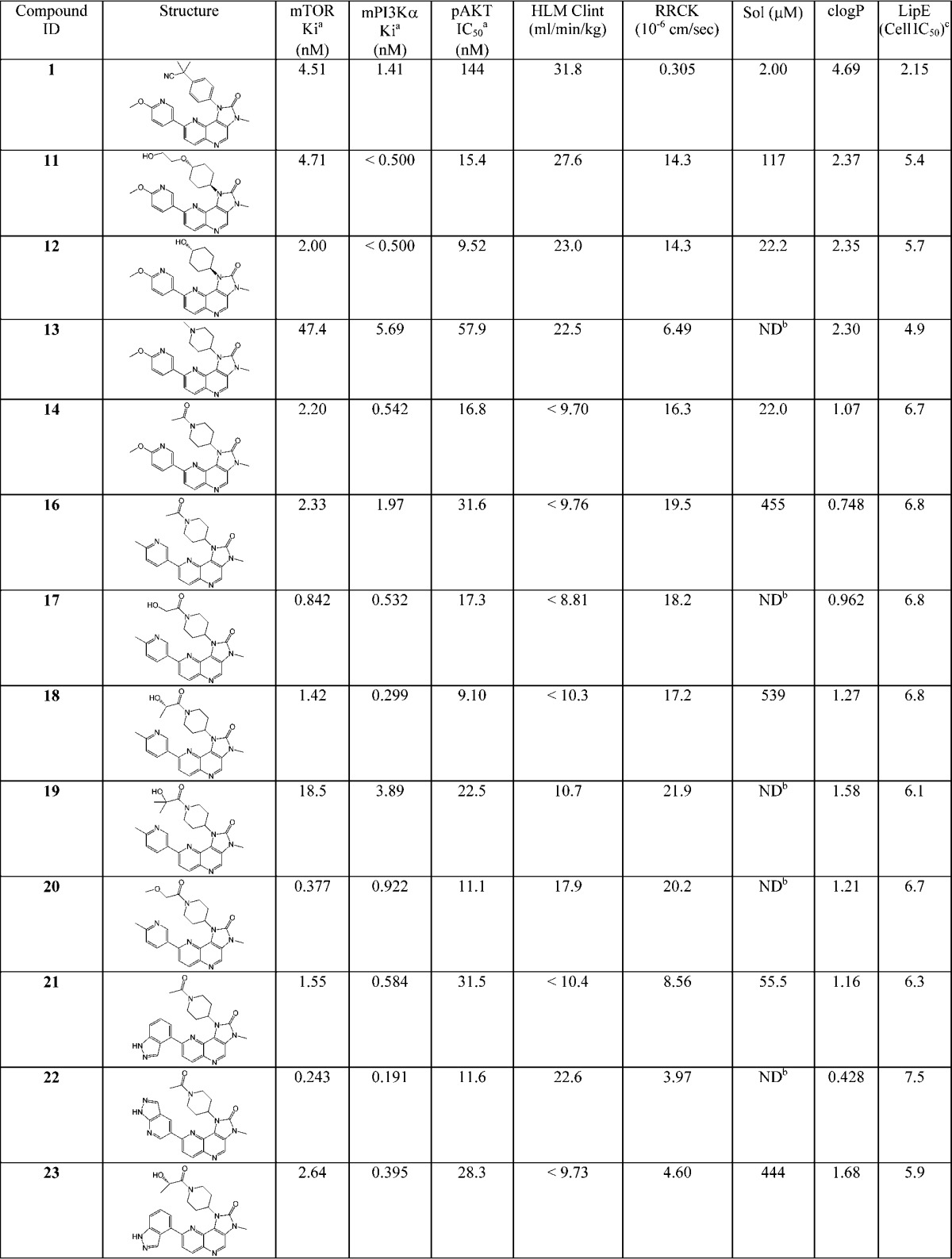
Compound 13 also exhibited relatively low intrinsic clearance in rat liver microsome assay (19.9 μL/min/mg). To determine the in vivo PK profile for a representative compound from the series, 13 was progressed to rat PK studies and exhibited favorable rat PK profile: Vdss = 4.46 L/kg, Cl = 24.2 mL/min/kg, T1/2 = 2.43 h, and F % = 76%.
Because 13 demonstrated favorable in vitro and in vivo ADMET properties, in vitro biotransformation of 13 was conducted in human liver S9. LC-MS/MS analysis showed that 13 was predominantly metabolized to 15 (Scheme 2). In addition, it was determined that oxidation of 13 to 15 did not require NADPH, a cofactor essential for P450-mediated oxidation, and the oxidation was readily inhibited by raloxifene, an aldehyde oxidase (AO) inhibitor.12 Together, these results indicated that AO was responsible for the oxidation of 13 to 15. After 15 was isolated form a large scale incubation of 13 with human S9, its structure was determined by 1H NMR analysis, confirming that the tricyclic core was the site of oxidation, consistent with the oxidative characteristics for AO.13 Because AO is a cytosolic enzyme, the stability of 13 was evaluated in a human liver S9 assay.14 Indeed, 13 was rapidly cleared in vitro in human liver S9 with a half-life of 6.5 min.
Scheme 2. AO Oxidation of 13.

Human PK prediction for P450-metabolized compounds has become a well-established and integrated process in lead optimization. However, for AO-metabolized compounds, human PK prediction remains a challenge because there is marked species differences in AO expression and there is no established in vitro and in vivo scaling method to calculate human clearance.14 Even though compound 13 exhibited a robust PK profile in rat, with its short half-life in human liver S9 assay, confidence for a robust human PK for 13 was low.
To select a compound with robust predicted human PK as a back-up to PF-04691502, a strategy to reduce the rate of AO oxidation to improve human PK was pursued. One way to reduce the AO oxidation rate is to disrupt the binding by increasing the bulkiness of the molecule.15 Compound 14 in Table 1 demonstrated the highest LipE and promising overall profile; hence, it was selected as the basis for the next round of designs.
Guided by SBDD, the methoxypyridine in 14 was replaced both by methylpyridine and by bicyclic heterocycles, and the acetyl group was substituted with an alcohol or an ether. These modifications would both increase bulkiness to reduce AO clearance and be tolerated with regard to in vitro potency because additional H bonds could be formed with the protein polar residues. Data for these designs, that is, compounds 16–23, are summarized in Table 1. These compounds exhibited good in vitro potency and good permeability. Low HLM clearance mediated by P450 was also observed for all of the compounds except compounds 20 and 22, which exhibited moderate clearance. As compared with 14, the follow-on designs also exhibited significantly increased solubility. The primary alcohol 17, the secondary alcohol 18, and the methylether 20 demonstrated increased potency against both mPI3Kα and mTOR. The tertiary alcohol 19 exhibited reduced binding affinity in the enzyme assays, probably due to unfavorable steric interactions between the bulky tertiary alcohol moiety and the proteins. Of the compounds containing bicyclic heterocycle side chains, 22 with the pyrazolopyridine side chain exhibited highly potent Ki against both mPI3Kα and mTOR. However, 22 showed low permeability and, consequently, a high ratio between cell IC50 and Ki.
Compound 18 demonstrated the best overall properties including the most potent cell IC50, low HLM clearance, high permeability, and very good solubility. A crystal structure of 18 bound with PI3Kγ was subsequently determined (Figure 3). The naphthyridine ring nitrogen of 18 bound in the hinge region, the methylpyridine nitrogen formed H bond with the conserved water molecule, and two additional H bonds were also formed between the amide carbonyl from 18 and Lys 833 side chain and between the alcohol from 18 and the Asp 964 side chain. The binding mode of 18 in PI3Kγ, determined by crystal structure, is consistent with the modeled binding mode of compound 1 in PI3Kγ.
Figure 3.
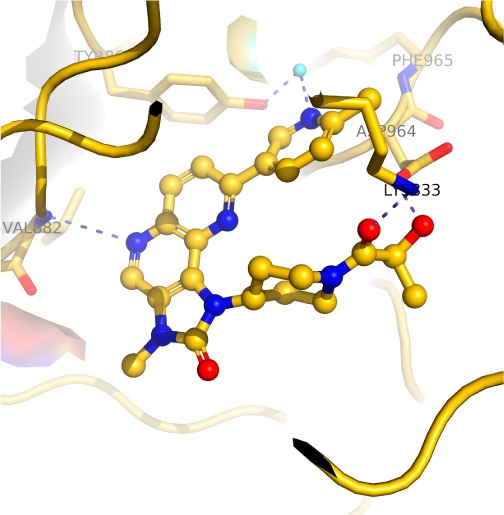
Cocrystal structure of 18 with PI3Kγ.
Six compounds from Table 1 were selected and tested in human liver S9 assay to determine their AO oxidation rate, and the half-lives are summarized in Table 2. For example, T1/2 for 18 is 38.7 min, which is significantly longer than that of 13. Furthermore, the indazole derivatives 21 and 23 are found to exhibit longer than 150 min half-lives. The data suggest that it is a validated strategy to reduce AO metabolism rate by increasing the size of the compounds to decrease their binding affinity with AO.
Table 2. AO Half Lives from in Vitro Human Liver S9 Assay.
| compd ID | human S9 T1/2 (min) | compd ID | human S9 T1/2 (min) |
|---|---|---|---|
| 16 | 8.60 | 19 | 12.2 |
| 17 | 21.9 | 21 | 158 |
| 18 | 38.7 | 23 | 156 |
With several tricyclic compounds that exhibited reduced AO clearance and met other project progression criteria, to prioritize them for in vivo studies, a strategy to predict their human clearance was developed. The conventional methods, including in vitro metabolic scaling and allometric scaling from preclinical species, have not successfully predicted human clearance for AO substrates. It was reported by Zientek14 that when in vitro S9 and cytosol stability data were used for metabolic scaling, the rank order of scaled intrinsic clearance was generally aligned between in vitro and in vivo data; however, the in vivo clearance for almost every compound evaluated was underestimated, with an average of 11-fold for the under estimation. Recently, Hutzler16a and Akabane16b reported a similar trend for metabolic scaling of AO substrates from stability data in cryopreserved human hepatocytes and custom pooled hepatocytes, respectively. An underestimation of 7.9–14.9-fold was reported by Akabane for the AO substrates.16b With AO clearance half-life determined for the tricyclic compounds from human liver S9 assay, we decided to build an empirical calibration curve for human clearance for AO substrates to describe the relationship between observed human clearance and clearance predicted by metabolic scaling method.8,8b Four known AO substrates with reported human pharmacokinetic data, namely, carbazeran,17 zoniporide,18 zaleplon,19 and PF-04217903,20 were selected to build an empirical calibration curve for human clearance for AO substrates to describe the relationship between observed human clearance and clearance predicted by metabolic scaling method.8,8b As shown in Table 3, these four compounds covered a broad range of stability (1–162 min) in human liver S9 in the presence of NADPH, which was included to capture both AO- and P450-mediated clearance in the incubations.
Table 3. Summary of Calculated Hepatic Plasma Clearance of AO Substrates in Human S9 as Compared with Reported Human Clearances.
| mL/min/kg |
||||||
|---|---|---|---|---|---|---|
| compd | T1/2 in S9 (NADPH) (min) | fu,p | blood/plasma ratio | fu,HS9 in silico | calcd CLH,Pa | obsd total CLPb |
| carbazeran | 1.01 | 0.08 | 0.70 | 0.735 | 10.6 | 38 |
| zoniporide | 20.8 | 0.34 | 0.75 | 0.753 | 5.1 | 21 |
| zaleplon | 64.3 | 0.49 | 0.74 | 0.548 | 3.7 | 16 |
| PF-04217903 | 162 | 0.16 | 0.90 | 0.912 | 0.40 | 6 |
Calculated CLH,P was calculated using the three equations described in the manuscript.
Observed intravenous plasma clearance except PF-04217903. The intravenous clearance of PF-04217903 was estimated based on observed oral clearance and physiological-based pharmacokinetic modeling.
To estimate the hepatic plasma clearance (CLH,P), the in vitro unbound intrinsic clearance (CLint,u) was first estimated from the in vitro human S9 stability data using eq 1, where t1/2 is the observed in vitro substrate depletion half-life (in min). As AO substrates were not stable in human S9, protein binding in human S9 could not be measured experimentally. Hence, protein binding of AO substrates was estimated by Pfizer in-house in silico models.
| 1 |
Human hepatic blood clearance (CLH,B) was calculated using CLint,u values and well-stirred model according to eq 2, where QH is human hepatic blood flow (21 mL/min/kg) and fu,B is fraction unbound in human blood. Hepatic plasma clearance (CLH,P) was calculated following eq 3.
| 2 |
| 3 |
Consistent with Zientek's finding, CLH,P calculated by metabolic scaling from in vitro data underestimated the observed human clearance for the four AO substrates tested (Table 3). Additional analysis was conducted to explore the relationship between the calculated and the observed clearances. As shown in Figure 4, a linear relationship was observed between calculated and observed plasma clearances for the four known AO substrates, which provided us an empirical scaling tool to predict human clearance for tricyclic compounds using in vitro human S9 stability data.
Figure 4.

Calibration curve of AO substrates between observed total CLP and calculated CLH,P based on in vitro stability data.
Human clearance was then predicted for the tricyclic derivatives by using the above-discussed linear calibration curve, and compounds 18 and 23 emerged as two leads with acceptable predicted human clearance, which is summarized in Table 4. These compounds were progressed to mouse PK and PK/PD studies. On the basis of the overall attributes including potency from in vitro biochemical mPI3Kα and mTOR assays and cellular assay, kinome selectivity, cerep broad ligand binding selectivity, predicted human clearance, and mouse PK/PD study results, 18 was selected to progress to mouse in vivo xenograft efficacy studies. As illustrated in Figure 5, 18 exhibited dose proportional tumor growth inhibition (TGI) in a U87MG mouse xenograft model, achieving 88% TGI at the highest tolerated dose, 40 mg/kg QD.21 Figure 6 shows that a robust in vivo PK/PD correlation for 18 was observed for samples from the high dose treatment group in the TGI studies.22 At 1 h time point, high free drug concentration in plasma yielded maximum inhibition of AKT phosphorylation. At 24 h time point, very low free drug concentration in plasma was correlated with minimum inhibition of AKT phosphorylation.6
Table 4. Predicted Human Clearance for the Top Two Tricyclic Derivatives.
| mL/min/kg |
||||||
|---|---|---|---|---|---|---|
| compd | T1/2 in S9 (NADPH) (min) | fu,p | blood/plasma ratio | fu,HS9 in silico | calcd CLH,Pa | predicted total CLP |
| 18 | 38.7 | 0.0875 | 0.86 | 0.670 | 1.1 | 8.1 |
| 23 | 156 | 0.101 | 1.01 | 0.593 | 0.38 | 5.8 |
Calculated CLH,P was calculated using the three equations described in the manuscript.
Figure 5.
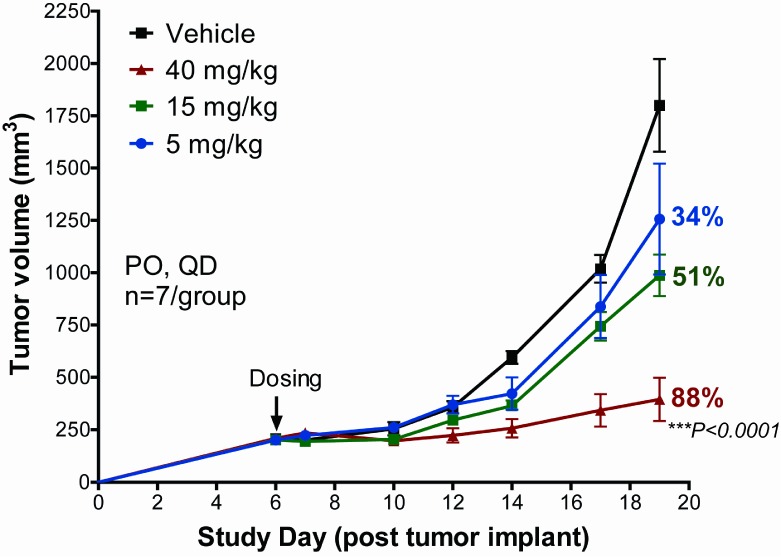
Tumor growth inhibition by 18 in U87MG mouse xenograft model.
Figure 6.
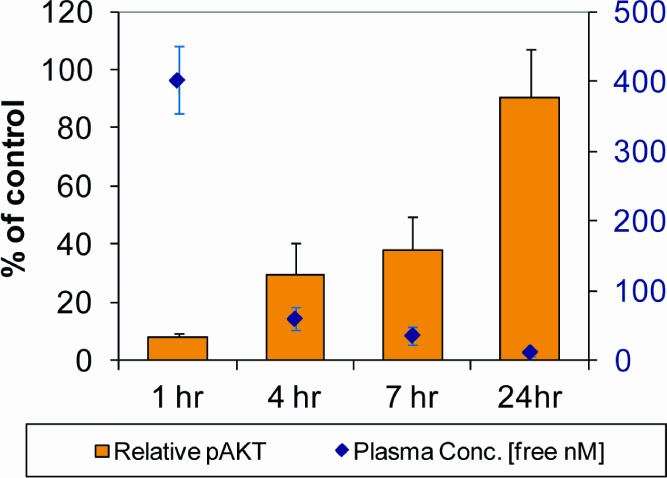
PK/PD correlation for 18.
Compound 18 was tested against human class I PI3K isoforms α, γ, and δ, with PI3Kα Ki of 0.130 nM, PI3Kγ Ki of 0.111 nM, and PI3Kδ Ki of 0.122 nM.23 Compound 18 was progressed to rat in vivo PK studies and exhibited robust PK profile: Vdss = 5.23 L/kg, Cl = 19.3 mL/min/kg, T1/2 = 1.85 h, and F % = 61%.
Shown in Figure 7 is a LipE plot to summarize the lead optimization process. In the plot, the X-axis represents clogP, the Y-axis represents p(cell IC50), and the markers are colored by RRCK value, wherein a blue color indicates RRCK > 5 × 10–6 cm/s, and red indicates RRCK value < 5 × 10–6 cm/s. In general, compounds with higher LipE are desired since they usually exhibit better in vivo properties.5 Compound 1 has high clogP, poor solubility, and high metabolic clearance. Through integration of SBDD and PPBO, 18 (PF-04979064) was discovered. As the spotfire plot illustrates, from compound 1 to PF-04979064, the LipE is increased by greater than 4 units. PF-04979064 exhibited excellent in vitro potency, very good solubility, high LipE, excellent kinome selectivity,24 robust PK/PD correlation and TGI efficacy, and acceptable predicted human clearance after incorporating both CYP- and AO-mediated metabolism. PF-04979064 was identified as a back-up candidate to PF-04691502.
Figure 7.
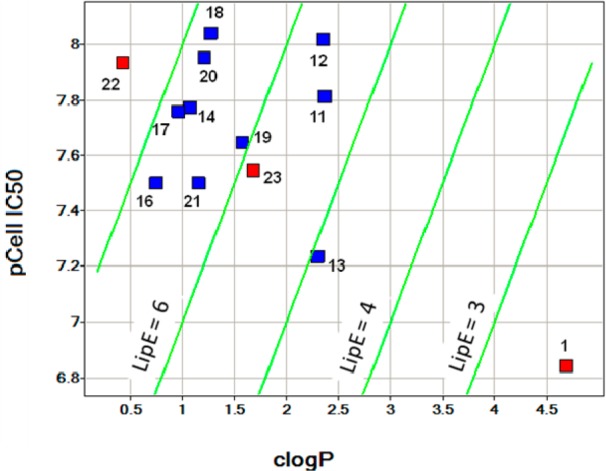
LipE plot to summarize the lead optimization.
Acknowledgments
Hieu Lam and Aihua Zou are acknowledged for biochemical screenings. Xiao-hong Yu is acknowledged for cellular screenings. Ya-Li Deng and Alexei Brooun are acknowledged for preparing crystals of PF-04979064 with PI3Kγ. Ketan Gajiwala is acknowledged for preparing Figure 3 and depositing the X-ray crystal structure. Jian Lin is acknowledged for running human liver cytosol assay for the selected tricyclic derivatives.25
Supporting Information Available
Invitrogen KSS data and % inhibition at 1 μM PF-04979064 tested at ATP concentration = Km. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The PDB accession code for the X-ray cocrystal structure of PI3Kγ + PF-04979064 is 4HVB.
The authors declare no competing financial interest.
Supplementary Material
References
- Eagleman J. A.; Luo J.; Cantley L. C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [DOI] [PubMed] [Google Scholar]
- Samuels Y.; Wang Z.; Bardelli A.; Silliman N.; Ptak J.; Szabo S.; Yan H.; Gazdar A.; Powell S. M.; Riggins G. J.; Willson J. K. V.; Markowitz S.; Kinzler K. W.; Vogelstein B.; Velculescu V. E. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [DOI] [PubMed] [Google Scholar]
- Shuttleworth S. J.; Silva F. A.; Cecil A. R. L.; Tomassi C. D.; Hill T. J.; Raynaud F. I.; Clarke P. A.; Workman P. Progress in the preclinical discovery and clinical development of class I and dual class I/IV phosphoinostide 3-kinase (PI3K) inhibitors. Curr. Med. Chem. 2011, 18, 2686–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney K. D.; Corcoran R. B.; Engelman J. A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2008, 28, 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Bagrodia S.; Bailey S.; Edwards M.; Hoffman J.; Hu Q.; Kania R.; Knighton D. R.; Marx M. A.; Ninkovic S.; Sun S.; Zhang E. Discovery of the Highly Potent PI3K/mTOR Dual Inhibitor PF-04691502 through Structure Based Drug Design. MedChemCommun. 2010, 1, 139–144Experimental procedures for in vitro biochemical assays and cellular assay were reported in this reference. [Google Scholar]
- Yuan J.; Mehta P. P.; Yin M.; Sun S.; Zou A.; Chen J.; Rafidi K.; Feng Z.; Nickel J.; Engebetsen J.; Hallin J.; Blasina A.; Zheng E.; Nguyen L.; Sun M.; Vogt P. K.; McHarg A.; Cheng H.; Christensen J. G.; Kan J. L. C.; Bagrodia S. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol. Cancer Ther. 2011, 10, 2189–2199Experimental procedures for the TGI study were reported in this reference. [DOI] [PubMed] [Google Scholar]
- Venkatesan A. M.; Dehnhardt C. M.; Delos Santos E.; et al. Bis morpholino 1,3,5-triazine derivatives: potent, ATP-competitive phosphatidylinositol-3-kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) inhibitors: discovery of PKI-587 a highly efficacious Dual Inhibitor. J. Med. Chem. 2010, 53, 2636–2645. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Bagrodia S.; Bailey S.. et al. Discovery of the potent PI3K/mTOR dual inhibitor PF-04979064. Oral presentation, MEDI-493, 240th ACS National Meeting, Boston, MA, United States, August 22–26, 2010.
- Cheng H.; Bailey S.; Baxi S. M.. et al. Structure-based drug design of PI3K/mTOR dual inhibitors and PI3K selective inhibitor from tricyclic imidazo[1,5]naphthyridine series. Poster presentation, 2011 AACR Special Conferences, Targeting PI3K/mTOR Signaling in Cancer, San Francisco, CA, United States, February 24–27, 2011.
- Maira S. M.; Stauffer F.; Brueggen J.; Furet P.; Schnell C.; Fritsch C.; Brachmann S.; Chene P.; De Pover A.; Schoemaker K.; Fabbro D.; Gabriel D.; Simonen M.; Murphy L.; Finan P.; Sellers W.; Garcia-Echeverria C. Identification and characterization of NVP-BEZ235, a new orally available dual PI3K/mTor inhibitor with potent in vivo antitumour activity. Mol. Cancer Ther. 2008, 7, 1851–1863The crystal structure of NVP-BEZ235 in PI3Kγ published in this paper was used as a reference for the initial docking studies for compound 1, using the Pfizer in-house 3D Molecular Visualization and Drug Design Tool. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Johnson T. W.; Hoffman J. E.; Guo L. C.; Liu Z.; Johnson T. O.; Liu K. K.. Preparation of imidazonaphthyridine derivatives for the use as PI3Kα inhibitors or PI3Kα/mTOR dual inhibitors, WO 2010038165 A1. Experimental procedures and NMR and MS data for all of the compounds discussed in this manuscript are included in the patent.
- Edwards M. P.Integration of Structure-based Drug Design and Physical Properties-based Optimization to Produce Cancer Clinical Candidates, 2010, AACR invited oral presentation, Washington, DC. [Google Scholar]
- Obah R. S. Potent inhibition of human liver aldehyde oxidase by raloxifen. Drug Metab. Dispos. 2004, 32, 89–97. [DOI] [PubMed] [Google Scholar]
- Garattini E.; Terao M. Increasing recognition of importance of aldehyde oxidase in drug development and discovery. Drug Metab. Rev. 2011, 43, 374–386. [DOI] [PubMed] [Google Scholar]
- Zientek M.; Jiang Y.; Youdim K.; Obach R. S. Drug Metab. Dispos. 2010, 38, 1322–1327The experimental procedure for the human liver S9 assay was reported in this paper. [DOI] [PubMed] [Google Scholar]
- Pryde D. C.; Tran T.; Jones P.; Duckworth J.; Howard M.; Gardner I.; Hyland R.; Webster R.; Wenham T.; Bagal S.; Omoto K.; Schneider R. P.; Lin J. Medicinal chemistry approaches to avoid aldehyde oxidase metabolism. Bioorg. Med. Chem. Lett. 2012, 22, 2856–2860. [DOI] [PubMed] [Google Scholar]
- Hutzler M.; Yang Y.; Albaugh D.; Fullenwider C. L.; Schmenk J.; Fisher M. B. Characterization of aldehyde oxidase enzyme activity in cryopreserved human hepatocyte. Drug Metab. Dispos. 2012, 40, 1322–1327. [DOI] [PubMed] [Google Scholar]
- Akabane T.; Gerst N.; Masters J. N.; Tamura K. A quantitative approach to hepatic clearance prediction of metabolism by aldehyde oxidase using custome pooled hepatocytes. Xenobiotics 2012, 42, 863–871. [DOI] [PubMed] [Google Scholar]
- Kaye B.; Offerman J. L.; Reid J. L.; Elliott H. L.; Hillis W. S. A species difference in the presystemic metabolism of carbazeran in dog and man. Xenobiotica 1984, 14, 935–945. [DOI] [PubMed] [Google Scholar]
- Dalvie. D.; Zhang C.; Chen W.; Smolarek T.; Obach R. S.; Loi C. M. Cross-species comparison of the metabolism and excretion of zoniporide: Contribution of aldehyde oxidase to interspecies differences. Drug Metab. Dispos. 2010, 38, 641–654. [DOI] [PubMed] [Google Scholar]
- Rosen A. S.; Fournie P.; Darwish M.; Danjou P.; Troy S. M. Zaleplon pharmacokinetics and absolute bioavailability. Biopharm Drug Dispos. 1999, 20, 171–175. [DOI] [PubMed] [Google Scholar]
- Data from Pfizer in-house file.
- U87MG tumor cells were subcutaneously implanted to nude mice, tumor-bearing mice were randomized and orally dosed once daily, and tumor sizes were measured and plotted. The tumor growth inhibition (TGI) was calculated as previously described6 and indicated on the graph for each treatment group. One-way ANOVA analysis was performed to compare treated groups to vehicle group. *** represents P < 0.0001.
- U87MG tumor-bearing mice were dosed with PF-04979064 at 40 mg/kg once, and tumors were harvested at various time points postdosing. Tumors then were processed, and tumor lysates were used to determine the inhibition of phosphorylation of AKT (pS473). Plasma was also collected, and drug concentration were analyzed. The inhibition of pAKT (left Y-axis) and corresponding plasma concentration of free drug (right Y-axis) were plotted against time points.
- Note: Compound 21 was not tested against human PI3Kβ. However, 21 was expected to inhibit human PI3Kβ equally potent as it inhibited other human class I PI3K isoforms.
- PF-04879064 was tested in a panel of 36 kinases at Invitrogen. At 1 μM, less than 25% inhibition was observed against all 36 kinases. Detailed kinome selectivity data for PF-04979064 can be found in the Supporting Information.
- AO clearance for the tricyclic compounds in Table 2 was also determined in the human liver cytosol assay. Human liver cytosol AO clearance data for the compounds are consistent with the clearance data from human liver S9 assay.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.