

Abstract
A series of 2-hydroxy-1,3-dioxoisoquinoline-4-carboxamides featuring an N-hydroxyimide chelating functionality was evaluated for their inhibitory properties against human immunodeficiency virus type 1 integrase (HIV-1 IN). Several derivatives displayed low nanomolar IC50 values comparable to that of the clinically used raltegravir. A marked effect of one compound on both primary IN-catalyzed reactions, strand transfer (ST), and 3′ processing (3′-P), emphasizes a novel IN inhibition mechanism establishing it as a potential new generation IN inhibitor. Substitution of the 2-hydroxyisoquinoline-1,3-dione scaffold at position 4 by carboxamido chains was beneficial for antiviral activity since reproducible low micromolar anti-HIV activities were obtained for the first time within this scaffold.
Keywords: HIV; antiretroviral; integrase; 3′ processing; 2-hydroxy-1,3-dioxoisoquinoline-4-carboxamide
HIV-1 integrase (IN) is a 32 kDa protein that plays a crucial role in HIV infection by incorporating the retrotranscribed viral DNA into the host chromosomal DNA. IN has been extensively studied as a therapeutic target in the field of AIDS antiretroviral therapy since it establishes irreversible infection and has no cellular equivalent, which limits toxicity.1,2 The integration process involves a sequence of 2 reactions, which both require the presence of metallic cofactors: in the cytoplasm, a DNA-IN complex is formed that catalyzes the endonucleolytic cleavage of a dinucleotide at each 3′-end of the dsDNA (the 3′-processing step, 3′-P) After transport into the nucleus, the strand transfer step (ST), catalyzed by the intasome (a specific tetramer of IN and viral DNA ends) then joins each 3′-end of this recessed DNA to a 5′-end in the host DNA.
The first FDA-approved drug acting as a strong selective ST inhibitor3 is raltegravir (Isentress). Elvitegravir, also recently approved, can be given once daily when combined with a booster (as part of the fixed-dose combination tablet Stribild),4 but cross-resistance rules out treatment of patients failing on raltegravir therapy.5,6 Dolutegravir (S/GSK1349572)7 is currently in phase III clinical trials. Although superior to raltegravir, it also exhibits significant resistance overlap.8 It can therefore be stated that IN still remains an orphan in terms of marketable drugs and an attractive and scientifically challenging target. One of the most innovative strategies so far was the design of inhibitors targeting the Lens Epithelium Derived Growth Factor (LEDGF)/p75 binding site on integrase (LEDGINs). These small molecules hinder the interaction of IN with the cellular cofactor LEDGF/p75. In addition LEDGINs stabilize integrase dimers and inhibit IN allosterically.9 Dual inhibitors against IN and reverse transcriptase (RT) have also been investigated. Very recently, a non-nucleoside pyrimidine-2,4-dione RT inhibitor was 3-N hydroxylated, leading to a N-hydroxyimide derivative capable of inhibiting IN.10
We previously designed and studied several series of 2-hydroxyisoquinoline-1,3-diones as potential dual inhibitors of HIV-1 IN and RT associated RNase H activities. A few hits displaying high selectivity for IN or the RT associated RNase H function with submicromolar IC50 values were discovered.11−13 Unfortunately nearly all tested compounds exhibited high cellular cytotoxicity in cell culture, which limited their applications as antiviral agents.
Careful examination of known potent IN strand transfer inhibitors (INSTIs) like raltegravir and dolutegravir points out the key presence of a magnesium chelating triad of oxygen atoms. Studies have also revealed the crucial importance of a hydrophobic side chain bearing a mono- or polyhalogenated benzyl group in the pharmacophore (Figure 1).6 With the publication of their PFV intasome crystal structures, Hare et al. recently demonstrated that this aromatic component invades a pocket at the protein–DNA interface, which is natively occupied by the 3′-terminal base of viral DNA.14 This led us to substitute our scaffold at position 4 with aromatic side chains, as exemplified by compound 33. This could enhance not only the magnesium-chelating properties of our molecules by extending the coplanar arrangement of oxygen atoms but also their affinity for the IN catalytic center through additional hydrophobic contacts, namely, π-stacking interactions with retroviral DNA bases.
Figure 1.
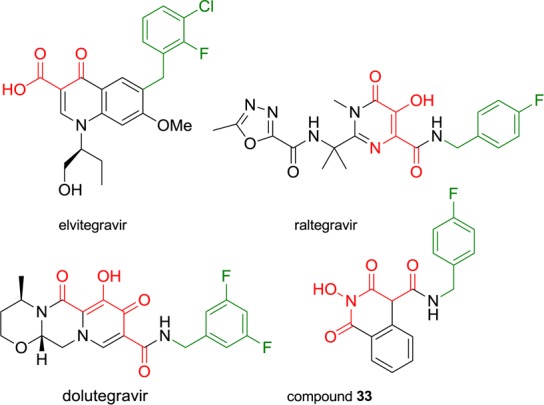
Structures of raltegravir, elvitegravir, dolutegravir, and compound 33 pointing out the key components of the HIV-1 IN inhibitory pharmacophore: the magnesium chelating moiety (red) and the hydrophobic halogenobenzyl group (green).
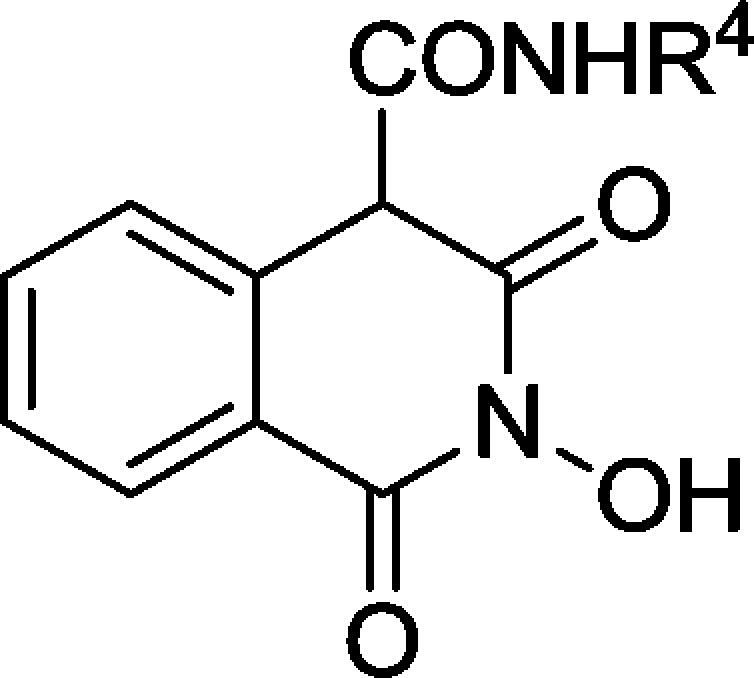
Herein, we propose further insight into the design of HIV IN inhibitors based on the 2-hydroxyisoquinoline-1,3-dione scaffold. The design, synthesis, and pharmacological evaluation of a novel series of 2-hydroxy-1,3-dioxoisoquinoline-4-carboxamides bearing diverse substituted phenyl, benzyl, or phenethyl side chains was carried out. The carboxamide link was chosen not only for its greater stability in physiological conditions, but also for its propensity to create an intramolecular hydrogen bond with the oxygen at position 3, thus orienting the aromatic side chain toward the desired hydrophobic cavity, as demonstrated by preliminary molecular modeling and docking studies (data not shown/see Supporting Information). Nonhydroxylated analogues 8 and 9 were also synthesized to validate the N-hydroxyl function as a key structural feature for magnesium complexation. The overall in vitro HIV-1 IN inhibitory potency, the anti-HIV activity in MT-4 cells, and cytotoxicity of the compounds were evaluated. Additionally, the in vitro inhibitions of HIV-1 IN 3′-P activity and RT associated RNase H function were also measured for the most interesting compounds.
The synthetic pathway was based upon a formerly reported route for the synthesis of 4-alkoxycarbonylisoquinoline-1,3-diones.15 Acid precursor 1 was obtained in two steps from commercial homophthalic acid. After coupling of O-benzylhydroxylamine, ammonia, or N-methylamine, the corresponding amides 2–4 were quantitatively cyclized in mild alkaline conditions into ester precursors 5–7 (Scheme 1). Diversification of the side chain was then obtained via addition–elimination of various primary or secondary amines on the ester moiety to afford protected compounds 8–21.The O-benzyl protecting group was finally removed with boron tribromide or boron trichloride to afford 2-hydroxy-1,3-dioxoisoquinoline-4-carboxamides 22–37. The target compounds were obtained either as the keto form or as a keto/enol mixture with a large preference for the keto form (measured by 1H NMR).
Scheme 1.

Table 1 reports the enzyme inhibitory activities of this series. First, it can be observed that compounds 8 and 9 are completely devoid of inhibitory activity against HIV-1 IN. This strongly suggests that the N-hydroxyl function participating in the chelating pharmacophore is indeed crucial to IN binding. The introduction of a phenylcarboxamido side chain (compounds 22–25) led to strong HIV-1 IN inhibition with IC50 values in the nanomolar range similar to that of raltegravir. The overall tendency seems to indicate that the linker’s length between the amide function and the aromatic ring does not have a drastic influence on activity since compounds 31, 32, and 33 bearing benzylcarboxamido side chains display IC50 values similar to those of their respective homologues 35, 36, and 34 with phenethylcarboxamido side chains. Even though halogen substitution of the aromatic ring seems favorable, its m,p-disubstitution with donating oxygenated substituents (−OH or -OMe) had a dramatic effect on the HIV-1 IN inhibition with a one-log collapse of the IC50 values (31, 32, 35, and 36) when compared to the o,p-disubstituted compounds 29 and 30. This may be explained by unfavorable steric hindrance preventing optimal π-stacking with the DNA cytosine in the hydrophobic cavity rather than by electronic effects. Moreover, replacing the amide proton by a methyl group (compound 37) provoked a 17-fold decrease of the HIV-1 IN inhibition, which stresses the importance of the side chain orientation and of the supposed intramolecular H-bond within the complex. Apart from overall HIV-1 IN inhibition, careful examination of the compounds’ effects on HIV-1 IN 3′-P and ST activities was also very instructive. A glance at the data of raltegravir evidence its well-known selectivity for ST over 3′-P, measured here at 128-fold. Close values of the IN overall and ST inhibitions were also observed for compounds 25–28 and 31–36, which may infer ST selectivity. In contrast, there was a discrepancy between overall IN and ST inhibition for compounds 23, 24, 29, and 30. The ST IC50 values are 10- to 15-fold higher than the overall inhibition, suggesting either a synergistic effect on both primary IN functions or the intervention of an unknown mechanism of IN inhibition. The 3′-P inhibition of compounds 23, 30, and 33 was thus examined, and the calculated ratios IC50(3′-P)/IC50(ST) were measured at 5.7, 4.1, and 0.7, respectively. Unlike raltegravir, ST selectivity is not retained for these isoquinoline-4-carboxamides, and to the best of our knowledge, compound 33 is the first low molecular IN inhibitor that displays inhibition of both IN catalytic steps in the low nanomolar range. We also tested some compounds against HIV-1 RT associated RNase H activity (Table 1), and submicromolar IC50 values were obtained for the most active compounds 29–30, 32–33, and 36.
Table 1. Inhibitory Activities of Compounds 8 and 9 and of the 2-Hydroxy-1,3-dioxoisoquinoline-4-carboxamides 22–37.

Concentration required to inhibit by 50% the in vitro overall, strand transfer, and 3′ processing integrase activities, respectively.
IC50 3′-P/IC50 ST ratio.
Concentration required to inhibit by 50% of the in vitro RNase H activity.
Finally, this series was evaluated in a cell-based antiviral assay against HIV-1 (Table 2). As expected, the non-N-2 hydroxylated compounds 8 and 9 were not active, suggesting that the antiviral potencies may strongly be related to the metal-chelating ability and integrase inhibition. The amide proton also proved essential for antiviral activity (EC50 > 107 μM for compound 37). Unfortunately, compounds 28, 31, 32, 35, and 36 were either poorly active or cytotoxic. Compounds 22–27, 33, and 34 ,however, displayed low micromolar EC50 values ranging from 1.75 to 5.08 μM. Although these are modest in comparison to that of the ST selective raltegravir (EC50 = 6.0 nM), it is still a very promising result since it is the first time that cytotoxicity is overcome on this scaffold. Compound 33 displayed a good aqueous solubility of 190.0 μM and a partition coefficient, log D of 0.56. At a 10 μM concentration, a mean permeability coefficient Papp (Caco-2 cells, pH 6.5/7.4) below 0.2 × 10–6 cm/s was determined, and high human plasma protein binding (mean of 99.9% protein bound) was observed, which might account for the lack of strong antiviral activity. This issue is currently being addressed through further pharmacomodulation. It must be stressed that, although we previously designed several compounds based on this scaffold, it is the first time that a reproducible low micromolar antiviral activity is attained in conjunction with a low nanomolar integrase inhibition. Moreover, compound 33 showed a profile similar to that of raltegravir and elvitegravir in time of addition experiments.24 This attests for the cellular inhibition of integrase by this scaffold, whereas inhibition of the RT associated RNase H function remained a side activity. Half of the series showed an advantageous window between antiviral efficacy and cellular toxicity (21- to 86-fold).
Table 2. Anti-HIV Activities of Compounds 8 and 9 and of the 2-Hydroxy-1,3-dioxoisoquinoline-4-carboxamides 22–37.
| compd | EC50a (μM) | CC50b (μM) | TIc |
|---|---|---|---|
| 8 | >250 | >250 | |
| 9 | >250 | >250 | |
| 22 | 4.95 | 105.5 | 21.3 |
| 23 | 3.34 | 12.3 | 3.7 |
| 24 | 2.47 | 64.0 | 25.9 |
| 25 | 1.75 | 114.5 | 65.4 |
| 26 | 3.12 | 130 | 41.7 |
| 27 | 5.7 | >125 | >22 |
| 28 | 17.63 | 118.5 | 6.7 |
| 29 | 9.24 | 60.4 | 6.5 |
| 30 | 7.94 | >125 | >16 |
| 31 | >125 | 125 | |
| 32 | >11 | 11 | |
| 33 | 2.34 | 202 | 86.3 |
| 34 | 5.08 | 123.5 | 24 |
| 35 | 70.77 | >125 | >1.8 |
| 36 | >63 | 63 | |
| 37 | >107.2 | 107.2 | |
| raltegravir | 0.006 | >8.0 | >1333 |
Effective concentration required to reduce HIV-1-induced cytopathic effect by 50% in MT-4 cells.
Cytotoxic concentration required to reduce MT-4 cell viability by 50%.
Therapeutic index, defined by CC50/EC50.
In silico docking studies were also performed in order to determine a possible binding mode with the target. Although our previously reported method was originally based on the PDB:3L2T crystallographic structure of PFV-IN intasome in complex with raltegravir,14 we decided to adapt it to the more recent 3S3M X-ray structure of the PFV intasome bound to dolutegravir (see Supporting Information).16 Whereas the invariant 3′-deoxyadenosine is flipped out of the active site in the case of elvitegravir and MK-0536, a raltegravir-derived INSTI with improved resistance profile,17 it seems to participate in additional π-stacking interactions with the core of dolutegravir in the 3S3M structure. In a similar manner, the extended planar aromatic nature of our N-hydroxyisoquinoline-1,3-dione core bearing the metal chelating pharmacophore infers a great propensity to interact with this 3′-deoxyadenosine via π-stacking contacts.
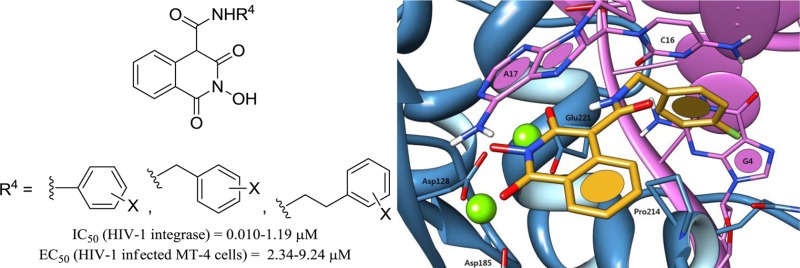
As expected, two possible binding modes were obtained for compound 33 using this model (Figure 2), both of which show similar statistical significance and high overall fitness function scoring. Both poses involve (a) dual magnesium complexation, (b) π-stacking of the fluorobenzyl side chain with the invariant deoxycytosine C16, and (c) π-stacking of the central isoquinoline moiety with the invariant terminal 3′-deoxyadenosine A17. Although pose 2B involving the exocyclic oxygen in the chelation pharmacophore is not to be excluded, we strongly think that pose 2A is more likely to occur in reality. A closer look at the weighed terms of the CHEMPLP fitness function indeed reveals that despite a slightly better metal chelation score, the ligand conformation in pose 2B requires significant internal torsion and close steric contacts in the carboxamide linkage. Conversely, not only does pose 2A allow a more favorable dihedral angle at this linkage but it also involves an additional intramolecular hydrogen bond between the amide proton of the 4-(4-fluorobenzylcarboxamido) side chain with the oxygen at position 3, which may direct and maintain the aromatic ring toward the desired hydrophobic pocket. If this docking model may only reflect the ST inhibition mechanics of our molecules, we cannot yet provide a theoretical explanation for the activity on 3′-processing.
Figure 2.

Putative binding modes of compound 33 in the PFV IN catalytic site obtained by molecular docking using the GOLD docking suite and the CHEMPLP fitness function. The ligand is depicted in orange, magnesium cations in green, IN in blue, and viral DNA in pink. Pose A: the three oxygens on the heterocyclic core contribute to Mg2+ chelation, allowing an intramolecular H-bond within the ligand. π-stacking interactions occur with deoxycytosine C16 and deoxyadenosine A17. Pose B: both π-stacking interactions occur as well. The exocyclic amide oxygen contributes to the metal chelation pharmacophore, at the expense of internal ligand torsion.
To our knowledge, it is the first time that such cumulative and synergistic effects on both integrase primary functions leading to strong integrase inhibition are observed. Little is known about 3′-P inhibition mechanism since only ST selective IN inhibitors have been cocrystallized with PFV IN so far. Crystal structures of PFV IN bound to unprocessed viral DNA prior to 3′-P were recently reported. As stipulated by Hare et al.,18 the selectivity of known IN inhibitors for ST may be explained by the fact that their binding to the catalytic site in pre-3′-P configuration would require the displacement of the 3′-terminal AAT trinucleotide, involving the rupture of phosphate–metal and phosphate–amide interactions, as opposed to the displacement of only one deoxyadenosine at the ST stage. Considering the apparent mobility of the unprocessed terminal 3′-dinucleotide in the catalytic site, such a mechanism might be envisaged. However, the energetic barrier needed for such a displacement might be difficult to reach, and a compound would need to establish additional contacts in the pre-3′-P complex in order to balance the energetics of binding and maintain its potency.
Compounds inhibiting 3′-P and ST catalytic activities in the same range are rare. They are exemplified by some conjugates of single-stranded oligonucleotides with hydrophobic molecules acting in the low nanomolar range19 and by low molecular compounds like numerous polyphenols, salicylhydrazides,20 pyranodipyrimidines,21 and styrylquinolines.22,23 The latter were very instructive, as a few compounds inhibited HIV-1 replication at low micromolar concentrations. After being characterized as metal chelating inhibitors, further cellular mechanistic investigations showed that they exert their IN inhibitory activity by interacting with the viral DNA or target DNA binding regions of IN and/or by interfering with the nuclear import mechanism of IN. In our case, the loss of the N-hydroxyl function strongly impaired inhibitory properties, suggesting that our compounds may target the HIV-1 IN catalytic site. However, at this stage, another mechanism of inhibition cannot entirely be ruled out. The alternative would consist of an allosteric mechanism through interactions with a specific region of IN prior to DNA binding. No effect of compound 33 was observed on IN/LEDGF binding (IC50IN/LEDGF ≫ 100 μM), excluding the interference with the cellular cofactor LEDGF/p75.
In summary, the introduction of a carboxamido side chain at position 4 of the 2-hydroxyisoquinoline-1,3-dione scaffold proved to be highly beneficial since low nanomolar anti-integrase activities close to that of raltegravir were obtained. In vitro enzymatic assays highlighted the unique behavior of these molecules: the typical ST selectivity of raltegravir and elvitegravir was not retained as a marked inhibition of the 3′-P step was observed. This pharmacomodulation had a positive effect on the antiviral activities as well since the limiting cytotoxicity of this scaffold was overcome and, for the first time, some compounds demonstrated low micromolar anti-HIV activities. Last but not least, compound 33 retained full activity against all common INSTI resistant strains tested (E92Q, Q148H, N155H, and G140S/Q148H) indicating a lack of cross-resistance with first-generation INSTIs and encouraging further clinical development.24
Glossary
Abbreviations
- HIV-1 IN
human immunodeficiency virus type 1 integrase
- ST
strand transfer
- 3′-P
3′ processing
- AIDS
acquired immune deficiency disease
- FDA
food and drug administration
- LEDGF
lens epithelium derived growth factor
- LEDGIN
inhibitor of the lens epithelium derived growth factor binding site in integrase
- RT
reverse transcriptase
- INSTI
integrase strand transfer inhibitor
- PFV
prototype foamy virus
Supporting Information Available
Synthesis of 8–9 and 22–37, experimental details, docking protocol, compound characterization (1H, 13C NMR, and HRMS) for final products, and biological assay methods. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was financially supported by grants from le Ministère de l’Enseignement Supérieur et de la Recherche Française, le Centre National de la Recherche Scientifique (CNRS), and l’Agence Nationale de la Recherche contre le Sida (ANRS). Experiments at KU Leuven were funded by the FP7 project CHAARM and the IWT.
The authors declare no competing financial interest.
Supplementary Material
References
- Ramkumar K.; Serrao E.; Odde S.; Neamati N. HIV-1 integrase inhibitors: 2007–2008 update. Med. Res. Rev. 2010, 30, 890–954. [DOI] [PubMed] [Google Scholar]
- McColl D. J.; Chen X. Strand transfer inhibitors of HIV-1 integrase: bringing IN a new era of antiretroviral therapy. Antiviral Res. 2010, 85, 101–118. [DOI] [PubMed] [Google Scholar]
- Summa V.; Petrocchi A.; Bonelli F.; Crescenzi B.; Donghi M.; Ferrara M.; Fiore F.; Gardelli C.; Paz O. G.; Hazuda D. J.; Jones P.; Kinzel O.; Laufer R.; Monteagudo E.; Muraglia E.; Nizi E.; Orvieto F.; Pace P.; Pescatore G.; Scarpelli R.; Stillmock K.; Witmer M. V.; Rowley M. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008, 51, 5843–5855. [DOI] [PubMed] [Google Scholar]
- Marchand C. The elvitegravir quad pill: the first once-daily dual-target anti-HIV tablet. Expert. Opin. Invest. Drugs 2012, 21, 901. [DOI] [PubMed] [Google Scholar]
- Shimura K.; Kodama E.; Sakagami Y.; Matsuzaki Y.; Watanabe W.; Yamataka K.; Watanabe Y.; Ohata Y.; Doi S.; Sato M.; Kano M.; Ikeda S.; Matsuoka M. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor Elvitegravir (JTK-303/GS-9137). J. Virol. 2008, 82, 764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrao E.; Odde S.; Ramkumar K.; Neamati N. Raltegravir, elvitegravir, and metoogravir: the birth of “me-too” HIV-1 integrase inhibitors. Retrovirology 2009, 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz J. C.; Rockstroh J. K. S/GSK1349572, a new integrase inhibitor for the treatment of HIV: promises and challenges. Expert Opin. Invest. Drugs 2011, 20, 537–548. [DOI] [PubMed] [Google Scholar]
- Canducci F.; Ceresola E. R.; Boeri E.; Spagnuolo V.; Cossarini F.; Castagna A.; Lazzarin A.; Clementi M. Cross-resistance profile of the novel integrase inhibitor dolutegravir (S/GSK1349572) using clonal viral variants selected in patients failing raltegravir. J. Infect. Dis. 2011, 204, 1811–1815. [DOI] [PubMed] [Google Scholar]
- Christ F.; Voet A.; Marchand A.; Nicolet S.; Desimmie B. A.; Marchand D.; Bardiot D.; Van der Veken N. J.; Van Remoortel B.; Strelkov S. V.; De Mayer M.; Chaltin P.; Debyser Z. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [DOI] [PubMed] [Google Scholar]
- Tang J.; Maddali K.; Dreis C. D.; Sham Y. K.; Vince R.; Pommier Y.; Wang Z. N-3 hydroxylation of pyrimidine-2,4-diones yields dual inhibitors of HIV reverse transcriptase and integrase. ACS Med. Chem. Lett. 2011, 2, 63–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billamboz M.; Bailly F.; Barreca M. L.; De Luca L.; Mouscadet J. F.; Calmels C.; Andréola M. L.; Christ F.; Debyser Z.; Witvrouw M.; Cotelle P. Design, synthesis and biological evaluation of a series of 2-hydroxyisoquinoline-1,3(2H,4H)-diones as dual inhibitors of human immunodeficiency virus type 1 integrase and reverse transcriptase RNase H domain. J. Med. Chem. 2008, 51, 7717–7730. [DOI] [PubMed] [Google Scholar]
- Billamboz M.; Bailly F.; Lion C.; Calmels C.; Andréola M. L.; Witvrouw M.; Christ F.; Debyser Z.; De Luca L.; Chimirri A.; Cotelle P. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as inhibitors of HIV-1 integrase and reverse transcriptase RNase H domain: influence of the alkylation of position 4. Eur. J. Med. Chem. 2011, 46, 535–546. [DOI] [PubMed] [Google Scholar]
- Billamboz M.; Bailly F.; Lion C.; Touati N.; Vezin H.; Calmels C.; Andréola M. L.; Christ F.; Debyser Z.; Cotelle P. Magnesium chelating 2-hydroxyisoquinoline-1,3-(2H,4H)-diones as inhibitors of HIV-1 integrase and/or the HIV-1 reverse transciptase ribonuclease H domain: discovery of a novel selective inhibitor of the ribonuclease H function. J. Med. Chem. 2011, 54, 1812–1824. [DOI] [PubMed] [Google Scholar]
- Hare S.; Gupta S. S.; Valkov E.; Engelman A.; Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billamboz M.; Bailly F.; Cotelle P. Facile synthesis of 4-alkoxycarbonylisoquinoline-1,3-diones and 5-alkoxycarbonyl-2-benzazepine-1,3-diones via a mild alkaline cyclization. J. Heterocyclic Chem. 2009, 46, 392–398. [Google Scholar]
- Hare S.; Smith S. J.; Métifiot M.; Jaxa-Chamiec A.; Pommier Y.; Hughes S. H.; Cherepanov P. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol. Pharmacol. 2011, 80, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métifiot M.; Johnson B.; Smith S.; Zhao X. Z.; Marchand C.; Burke T.; Hughes S.; Pommier Y. MK-0536 inhibits HIV-1 integrases resistant to raltegravir. Antimicrob. Agents Chemother. 2011, 55, 5127–5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Maertens G. N.; Cherepanov P. 3′-Processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 2012, 3020–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agapkina J.; Zatsepin T.; Knyazhanskaya E.; Mouscadet J. F.; Gottikh M. Structure–activity relationship studies of HIV-1 integrase oligonucleotide inhibitors. ACS Med. Chem. Lett. 2011, 2, 532–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mawsawi L. Q.; Dayam R.; Taheri L.; Witvrouw M.; Debyser Z.; Neamati N. Discovery of novel non-cytotoxic salicylhydrazide containing HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6472–6475. [DOI] [PubMed] [Google Scholar]
- Witvrouw M.; Van Maele B.; Vercammen J.; Hantson A.; Engelborghs Y.; De Clercq E.; Pannecouque C.; Debyser Z. Novel inhibitors of HIV-1 integration. Curr. Drug Metab. 2004, 5, 291–304. [DOI] [PubMed] [Google Scholar]
- Deprez E.; Barbe S.; Kolaski M.; Leh H.; Zouhiri F.; Auclair C.; Brochon J. C.; Le Bret M.; Mouscadet J. F. Mechanism of HIV-1 integrase inhibition by styrylquinoline derivatives in vitro. Mol. Pharmacol. 2004, 65, 85–98. [DOI] [PubMed] [Google Scholar]
- Mousnier A.; Leh H.; Mouscadet J. F.; Dargemont C. Nuclear import of HIV-1 integrase is inhibited in vitro by styrylquinoline derivatives. Mol. Pharmacol. 2004, 66, 783–788. [DOI] [PubMed] [Google Scholar]
- Desimmie B. A.; Demeulemeester J.; Suchaud V.; Taltynova O.; Billamboz M.; Lion C.; Bailly F.; Strelkov S. V.; Debyser Z.; Cotelle P.; Christ F. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones (HIDs), novel inhibitors of HIV integrase with a high barrier to resistance. ACS Chem. Biol. 2013, 10.1021/cb4000426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.