

Abstract
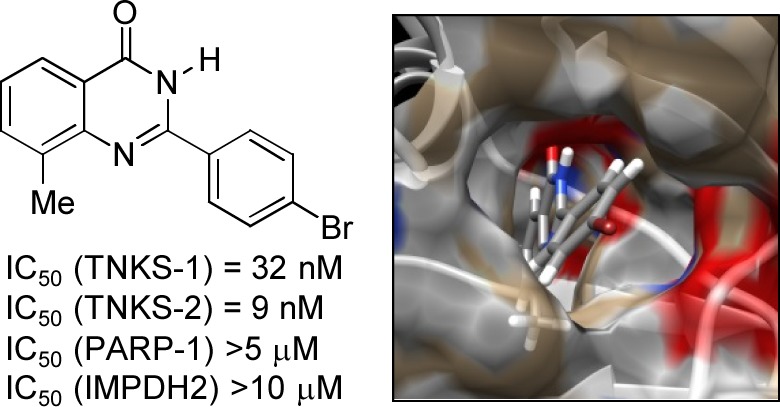
Tankyrases (TNKSs) are poly(ADP-ribose)polymerases (PARPs) that are overexpressed in several clinical cancers. They regulate elongation of telomeres, regulate the Wnt system, and are essential for the function of the mitotic spindle. A set of 2-arylquinazolin-4-ones has been designed and identified as potent and selective TNKS inhibitors, some being more potent and selective than the lead inhibitor XAV939, with IC50 = 3 nM vs. TNKS-2. Methyl was preferred at the 8-position and modest bulk at the 4-position of the 2-phenyl group; electronic effects and H-bonding were irrelevant, but charge in the 4′-substituent must be avoided. Molecular modeling facilitated initial design of the compounds and rationalization of the SAR of binding into the nicotinamide-binding site of the target enzymes. These compounds have potential for further development into anticancer drugs.
Keywords: Tankyrase, quinazolin-4-one, PARP, selectivity, hydrophobic pocket
Tankyrase-1 (PARP-5a, TNKS-1, ARTD5) and tankyrase-2 (PARP-5b, TNKS-2, ARTD6) are members of the poly(ADP-ribose)polymerase (PARP) superfamily of enzymes. PARPs transfer ADP-ribose units from substrate NAD+ to acceptor proteins, building poly(ADP-ribose) polymers.1 TNKS-1 was first reported in 1998;2 it is responsible for regulating the (re)elongation of telomeres through poly(ADP-ribosyl)ation of telomere repeating binding factor-1 (TRF-1) and autopoly(ADP-ribosyl)ation.3,4 Tankyrases have been proposed to be therapeutic targets in cancer on the basis of this role at telomeres, leading the cells into telomere-shortening crisis, much in the manner of telomerase inhibitors.5 Tankyrase-1 is also required for correct structure and function of the mitotic spindle and centrosome, where the main protein substrate is NuMA.6 Aberrant Wnt signaling is involved in many human cancers, and inhibitors of this system show antitumor activity in vivo.7,8 The specific protein target for poly(ADP-ribosyl)ation by tankyrases in the Wnt system is axin, stimulating its degradation through the ubiquitin-proteasome pathway. Thus, inhibition of tankyrases results in prolonged life of axin and increases the amount of the destruction complex of β-catenin. This leads to decreased levels of β-catenin and increased levels of phosphorylated β-catenin causing inhibition of the Wnt/β-catenin-driven proliferation of cancer cells.9
Tankyrases are overexpressed in several human cancers, including breast cancer,10 colon cancer,11 chronic myeloid leukemia,12 brain tumors,13 and gastric14 and bladder cancers.15 There is 85% sequence homology between TNKS-1 and TNKS-2 within the ankyrin repeat, sterile alpha motif, and the NAD+-binding catalytic domains; the catalytic domains are 86% identical. TNKS-1 contains an N-terminal HPS domain, which contains homopolymeric tracts of His, Pro, and Ser; this is absent from TNKS-2.2,16 Some of their functions are specific to one or other isoform, but the two tankyrases can act redundantly for many. Thus, in designing a therapeutic inhibitor, selectivity with respect to other NAD+-binding enzymes (e.g., other PARPs and oxidoreductases) is desirable but selectivity between the tankyrases is not.
A few tankyrase inhibitors have been reported to date. XAV939 1 (Figure 1) was identified by high-throughput screening as an inhibitor of Wnt signaling; it was subsequently confirmed that this effect was due to potent and selective inhibition of the tankyrases.9 This compound is reported to show IC50 vs TNKS-1 = 11 nM and IC50 vs TNKS-2 = 4 nM, in contrast to IC50 = 2.3 μM for inhibition of PARP-1. This compound has since been used as a molecular tool to study the Wnt pathway and its role in cancer and other cellular functions.17−20 Inhibitors of varying potency have also been identified, which bind at the adenosine-binding and other sites.21−23 Very recently, some quinazolin-4-ones carrying (CH2)2CONHAr at position-2 have been reported as binding both to the nicotinamide-binding site and at an induced pocket.24
Figure 1.
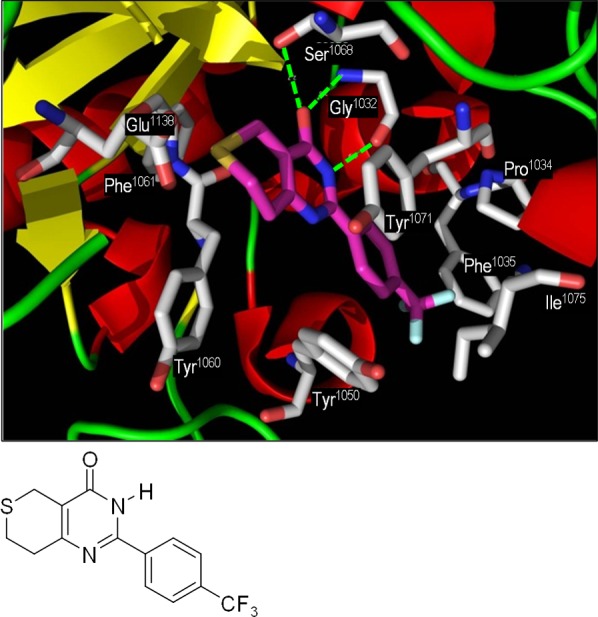
Structure of XAV939 1 and representation of the X-ray crystal structure of 1 bound into the NAD+-binding catalytic domain of human tankyrase-2.25 Key protein residues are in gray, and key H-bond interactions are shown as green dashed lines.
A crystal structure of 1 bound into the nicotinamide-binding region of the catalytic domain of TNKS-2 was published in 2010,25 revealing the classical PARP motif of H-bonding of the lactam carbonyl O with OH of Ser1068 and with NH of Gly1032 and of the lactam NH with C=O of Gly1032. Further π-stacking interactions are made from the lactam to Tyr1071, and the side-chain trifluoromethylphenyl projects into a hydrophobic pocket, π-stacking with Tyr1050 and making hydrophobic interactions with Pro1034, Phe1035, and Ile1075. Our interpretation of the binding of 1 in the crystal of tankyrase-2 is shown in Figure 1.
Using 1 as a structural lead and the above crystal structure25 as a basis for structure-based design, we designed a series of 2-arylquinazolin-4-ones as potential inhibitors of the tankyrases. In our target quinazolinones, the 2-aryl group is variously substituted at the 4-position, as the modeling guided us that the benzene ring would fit well into a hydrophobic pocket lined with Tyr1050, Tyr1060, and Pro1034. In this pose, the 4-substitutent on the benzene ring projects into a smaller hydrophobic tunnel, flanked by Phe1035 and Ile1075. The model showed that substitution would not be tolerated at the 5-, 6-, and 7-positions of the quinazolin-4-one but that small groups at the 8-position may be acceptable. Clearly, the lactam is designed for H-bonding to Gly1032 and Ser1068.
The quinazolin-4-ones were prepared by classical methods (Scheme 1). 2-(4-Chlorophenyl)quinazolin-4-one 3 was synthesized in moderate yield in one pot by condensation of anthranilamide 2 with 4-chlorobenzaldehyde in the presence of hydrogensulfite ion, with air oxidation of the intermediate aminal occurring in situ. The corresponding 3-methyl- and 3-methoxyanthranilamides 5a,b were prepared by reaction of the acids 4a,b with carbonyldiimidazole, followed by aqueous ammonia.26 Acylation of 5a,b with a range of 4-substituted benzoyl chlorides furnished the 3-methyl-2-aroylaminobenzamide derivatives 6a,b in excellent yields. Finally, cyclization of 6a,b under basic conditions provided 7a–i in good to excellent yields. The para-substituents on the 2-phenyl ring of quinazolin-4-ones 7a–i range from electron-donating to electron-withdrawing but lack potential for H-bond donation. Transfer hydrogenation of the nitro group of 7d gave the primary aniline of 7j. Demethylation of the methoxyphenyl group of 7e provided the corresponding phenol in 7k. To test the acceptability of a H-bond donor at the 8-position, 7i was similarly demethylated to afford the 8-hydroxyquinazolin-4-one 7l. To challenge the dimensions of the hydrophobic pocket, the 4-substituent of the phenyl ring was extended in 10. 4-Aminomethylbenzoic acid 8 was acylated at N with benzyl chloroformate under Schotten–Baumann conditions, before formation of the acyl chloride. This was used to acylate the anthranilamide 5a, providing the intermediate amide 9. The usual cyclization conditions of hot aqueous sodium hydroxide merely hydrolyzed the carbamate, but treatment with the weaker base potassium carbonate cyclized 9 smoothly to give 10. Removal of Cbz with HBr gave 11, which tested the tolerance of a basic amine (i.e., cation) in the 4′-position.
Scheme 1. Syntheses of Target 2-Arylquinazolin-4-ones 3, 7, and 11 and Locant Numbers.

Reagents and conditions: i, PhCHO, NaHSO3, AcNMe2, 150 °C, open flask; ii, CDI; iii, aq. NH3; iv, ArCOCl, pyridine; v, aq. NaOH (0.5 M), 60°C; vi, +NH4HCO2–, Pd/C, MeOH, DMF; vii, BBr3, CH2Cl2 reflux; viii, CbzCl, aq. K2CO3; ix, SOCl2; x, 5a, pyridine; xi, aq. K2CO3 (1.0 M), 100°C; xii, HBr, AcOH.
Compounds 3, 7f, 7i, and 7l test the required nature of the 8-substituent (H, Me, OMe, and OH, respectively), while keeping the 2-aryl group constant as 4-chlorophenyl. Similarly, the requirements for the 4-substituent are explored while keeping the 8-substituent constant as methyl (in 7a–l, 10, and 11). The members of this library were then evaluated for potency of inhibition of the catalytic activities of human TNKS-1 and TNKS-2, with counterscreening for selectivity against PARP-1 and a representative NAD+-requiring oxidoreductase, inosine monophosphate hydrogenase-2 (IMPDH2, itself the target of the active metabolite TAD of the experimental agent tiazofurin27 and metabolites of therapeutic 6-thioguanine and 6-mercaptopurine28).
Compounds were evaluated for inhibition of autopoly(ADP-ribosyl)ation activity of TNKS-2 using an assay developed in house. Full details are given in the Supporting Information. Compounds 3, 7f, 7i, and 7l, which differ only in the 8-substituent on the quinazolin-4-one core, were assessed for inhibition at one concentration, 10 nM. These data (3, 2 ± 5%; 7f, 49 ± 14%; 7i, 25 ± 5%; 7l, 32 ± 2% inhibition) showed that methyl is the 8-substituent giving the greatest potency. Further detailed studies were conducted keeping the 8-methyl group constant, while varying the 4-substituent on the 2-aryl group. IC50 values were then determined for inhibition of TNKS-2 by the focused library of 8-methylquinazolin-4-ones 7a–h,j,k, 10, and 11. These compounds were also examined for their ability to inhibit the activity of the isoform TNKS-1, using a commercial assay in which the enzyme transfers biotinylated ADPr units to immobilized histone-1. Two counterscreens were also used to assess the selectivity of inhibition of the tankyrases by the test compounds: PARP-1 and IMPDH2. The data are presented in Table 1.
Table 1. IC50 Values for Inhibition of Enzyme Activity by Test 2-Arylquinazolin-4-ones.
| compd | TNKS-1 IC50 (nM) | TNKS-2 IC50 (nM) | PARP-1 IC50 (nM) | IMPDH2 IC50 (μM) |
|---|---|---|---|---|
| 1 | 17 | 9.6 | 1200 | >10 |
| 7a | 42 | 41 | 795 | >10 |
| 7b | 35 | 6.7 | 685 | >10 |
| 7c | 48 | 10 | >5000a | >10 |
| 7d | 32 | 30 | 104 | >10 |
| 7e | 37 | 12 | 891 | >10 |
| 7f | 36 | 33 | >5000a | >10 |
| 7g | 32 | 5.5 | >5000a | >10 |
| 7h | 44 | 40 | >5000a | >10 |
| 7j | 35 | 3.3 | 504 | >10 |
| 7k | 52 | 5.5 | 1200 | >10 |
| 10 | 73 | 44 | >5000a | >10 |
| 11 | NDb | ∼100 | NDb | NDb |
Limited by solubility.
Not determined.
The standard inhibitor 1 showed IC50 values comparable with those reported9 using a different assay. We determined 1 to be 71-fold selective for inhibition of TNKS-1 and 133-fold selective for TNKS-2, both vs. PARP-1. Our 2-aryl-5-methylquinazolin-4-ones all showed potent and selective inhibition of the tankyrases. Compounds 7a–h,j,k were marginally less potent against TNKS-1 than was 1, with IC50 values very similar to each other at ca. 40 nM. These compounds were generally more potent against TNKS-2, with 7b (4′-Me), 7g (4′-Br), 7j (4′-NH2), and 7k (4′–OH) having lower IC50 values than did 1. Some structure–activity relationship (SAR) inferences can be drawn for the 4′-position. Very small substituents (H in 7a, F in 7h) reduce the activity significantly, with smaller effects of Cl in 7d and NO2 in 7f. The most potent compounds 7b,c,e,g,j,h carry similarly sized substituents; size appears to be much more important than electronic or polar/H-bonding properties (the aniline in 7j and the phenol in 7k would not be ionized). Interestingly, increasing the size of the 4′-substituent greatly, in 10, only results in slight loss of potency against TNKS-2 (10, IC50 = 44 nM; cf., 7k, IC50 = 3 nM, and 7a, IC50 = 41 nM). However, the primary aliphatic amine in 11, which would be protonated and cationic at the pH of the assay, caused major loss of activity against TNKS-2.
The counterscreen against PARP-1 used a commercial kit (Trevigen). All the 2-aryl-8-methylquinazolin-4-ones showed modest inhibition of PARP-1, up to the limit of solubility (Table 1). Some inhibitors showed equivalent or improved selectivity for tankyrases over PARP-1, compared with 1. For example, the 4′-CF3 analogue 7c was >100-fold and >500-fold selective for TNKS-1 vs. PARP-1 and TNKS-2 vs PARP-1, respectively; 7g (4′-Br) was >150-fold and >830-fold selective, respectively; 7j (4′-NH2) was 14-fold and 170-fold selective, respectively; 7k (4′-OH) was 23-fold and 240-fold selective, respectively. Even 10, with the very long 4′-group, showed good selectivity (>70-fold TNKS-1 vs. PARP-1, >110-fold TNKS-2 vs. PARP-1). These modest activities vs. PARP-1 correlate well with data for 7a,c–e,j,k reported by Griffin et al.29 who investigated quinazolin-4-ones as PARP-1 inhibitors.
The known inhibitor 1 and our new potent tankyrase inhibitors were counterscreened against IMPDH2, an oxidoreductase that uses NAD+, to examine selectivity further. No inhibitory activity was seen for 1 and for 7a–h,j,k and 10 up to 10 μM (Table 1), indicating extremely high selectivity for these compounds for the tankyrases over IMPDH2. This is expected, as tiazofurin itself is inactive but must be metabolized to TAD for potent inhibition.27
To aid in understanding of the mode of binding and of the SARs, the structures of selected quinazolin-4-ones were docked into the nicotinamide-binding site of tankyrase-2 (containing 1), using the published crystal structure.25 Structures of the quinazolin-4-ones were minimized and charged using Sybyl and were docked onto the protein using the lactam as a fixed feature. The ligands were subjected to molecular dynamics and molecular mechanics optimization. The upper four panels of Figure 2 show the structures of four 2-(4-chlorophenyl)quinazolin-4-ones with different 8-substituents bound into this site. In 7f (8-Me), 7i (8-OMe), and 7l (8-OH), the small 8-substituents project into a small pocket in the protein surface, whereas this pocket is unoccupied when 3 (8-H) is bound. The methoxy group in 7i is carefully angled to project the methyl further into this pocket. These observations explain the weaker activity of 3 at 10 nM, compared with 7f,i,l.
Figure 2.
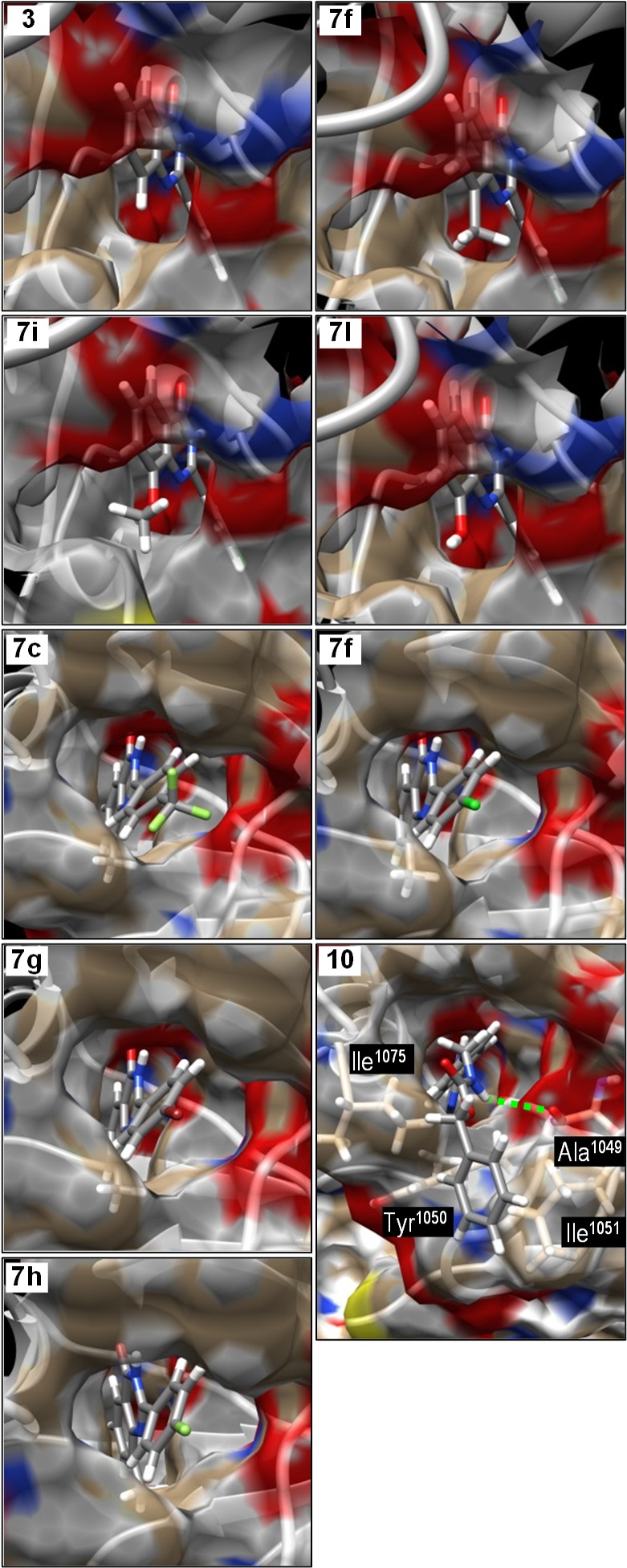
Molecular modeling of 2-arylquinazolin-4-one inhibitors into the structure of the nicotinamide-binding site of tankyrase-2. The upper four panels compare the binding of the 8-substituent while keeping the 2-aryl group constant. The lower five panels show the location of the 2-aryl group in the hydrophobic pocket, placing the 4′-substituent in a narrower tunnel through to solvent water.
The lower five panels of Figure 2 show the binding of five quinazolin-4-ones with the same 8-substitutent (Me) but with five different 4′-substituents on the 2-phenyl group. The 2-aryl groups project into the center of a large hydrophobic pocket, lined with aromatic residues. On the opposite face of the pocket, there is a narrower hydrophobic tunnel leading through to solvent water; the lower five panels of Figure 2 show the views from the exterior through the tunnel into the hydrophobic pocket. The 2-phenyl fits the large pocket snugly; the small 4′-substituents are then located in the inner end of the tunnel. This local environment tolerates moderately narrow substituents, irrespective of polarity or H-bonding, but its lining is intolerant of the highly polar cation in 11. Most interesting is the binding of 10. The long CbzNHCH2 group threads through the tunnel, placing the remote benzene ring in solvent where it interacts with hydrophobic residues Ile1075, Tyr1050, and Ile1051 on the protein surface. The CbzN–H makes a partly occupied H-bond (2.9 Å O–N) with Ala1052 carbonyl in the tunnel, pointing to further opportunities for design of second-generation inhibitors.
Here, we report the design and early evaluation of a series of 2-arylquinazolin-4-ones as potent and highly selective inhibitors of the tankyrases, important new target enzymes for cancer treatment. Methyl at the 8-position appears to be optimum for potency, and a variety of substituents are tolerated at the para-position of the 2-phenyl ring. These quinazolin-4-ones are highly selective for inhibition of tankyrases-1 and -2, showing much weaker inhibition of the most abundant PARP, PARP-1, and no effect whatsoever on an NAD+-requiring oxidoreductase, IMPDH2. Molecular modeling revealed the structural basis of potency and selectivity in which the 2-aryl group was accommodated in a hydrophobic pocket from which a narrow tunnel leads to the solvent. These compounds provide important leads for design and development of pharmaceutically useful drugs for treatment of cancer and other diseases.
Acknowledgments
We thank Dr. Lari Lehtiö (University of Oulu) for helpful discussions.
Glossary
ABBREVIATIONS
- ADP
adenosine-diphosphate-ribose
- Cbz
benzyloxycarbonyl
- IMPDH
inosine monophosphate dehydrogenase
- NAD+
nicotinamide adenine dinucleotide
- PARP
poly(ADP-ribose)polymerase
- TAD
tiazofurin adenine dinucleotide
- TNKS
tankyrase
- TRF-1
telomere repeating binding factor-1
Supporting Information Available
Details of chemical syntheses, characterization of inhibitors, biochemical methods and IC50 curves, etc. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors thank the Association for International Cancer Research for generous financial support.
The authors declare no competing financial interest.
Supplementary Material
References
- Javle M.; Curtin N. J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.; Giriat I.; Schmitt A.; de Lange T. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 1998, 282, 1484–1487. [DOI] [PubMed] [Google Scholar]
- Cook B. D.; Dynek J. N.; Chang W.; Shostak G.; Smith S. Role for the related poly(ADP-ribose) polymerases tankyrase 1 and 2 at human telomeres. Mol. Cell. Biol. 2002, 22, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seimiya H.; Muramatsu Y.; Smith S.; Tsuruo T. Functional subdomain in the ankyrin domain of tankyrase 1 required for poly(ADP-ribosyl)ation of TRF1 and telomere elongation. Mol. Cell. Biol. 2004, 24, 1944–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A.; de Lange T. Regulation of telomerase by telomeric proteins. Annu. Rev. Biochem. 2004, 73, 177–208. [DOI] [PubMed] [Google Scholar]
- Chang P.; Coughlin M.; Mitchison T. J. Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat. Cell Biol. 2005, 7, 1133–1139. [DOI] [PubMed] [Google Scholar]
- Polakis P. Drugging Wnt signalling in cancer. EMBO J. 2012, 31, 2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waaler J.; Machon O.; Tumova L.; Dinh H.; Korinek V.; Wilson S. R.; Paulsen J. E.; Pedersen N. M.; Eide T. J.; Machonova O.; Gradl D.; Voronkov A.; von Kries J. P.; Krauss S. A novel tankyrase inhibitor decreases canonical wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012, 72, 2822–2832. [DOI] [PubMed] [Google Scholar]
- Huang S.-M. A.; Mishina Y. M.; Liu S.; Cheung A.; Stegmeier F.; Michaud G.; Charlat O.; Wiellette E.; Zhang Y.; Wiessner S.; Hild M.; Shi X.; Wilson C. J.; Mickanin C.; Myer V.; Fazal A.; Tomlinson R.; Serluca F.; Shao W.; Cheng H.; Shultz M.; Rau C.; Schirle M.; Schlegl J.; Ghidelli S.; Fawell S.; Lu C.; Curtis D.; Kirschner M. W.; Lengauer C.; Finan P. M.; Tallarico J. A.; Bouwmeester T.; Porter J. A.; Bauer A.; Cong F. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 624–620. [DOI] [PubMed] [Google Scholar]
- Gelmini S.; Poggesi M.; Distante V.; Bianchi S.; Simi L.; Luconi M.; Casini Raggi C.; Cataliotti L.; Pazzaglia M.; Orlando C. Tankyrase, a positive regulator of telomere elongation, is over expressed in human breast cancer. Cancer Lett. 2004, 216, 81–87. [DOI] [PubMed] [Google Scholar]
- Shebzukhov Y. V.; Lavrik I. N.; Karbach J.; Khlgatian S. V.; Koroleva E.; Belousov P. V.; Kashkin K. N.; Knuth A.; Jager E.; Chi N.-W.; Kuprash D. V.; Nedospasov S. A. Human tankyrases are aberrantly expressed in colon tumors and contain multiple epitopes that induce humoral and cellular immune responses in cancer patients. Cancer Immunol. Immunother. 2008, 57, 871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell L. J.; Fidler C.; Eagleton H.; Peniket A.; Kusec R.; Gal1 S.; Littlewood T. J.; Wainscoat J. S.; Boultwood J. hTERT, the catalytic component of telomerase, is downregulated in the haematopoietic stem cells of patients with chronic myeloid leukaemia. Leukemia 2006, 20, 671–679. [DOI] [PubMed] [Google Scholar]
- Shervington A.; Patel R.; Lu C.; Cruickshanks N.; Lea R.; Roberts G.; Dawson T.; Shervington L. Telomerase subunits expression variation between biopsy samples and cell lines derived from malignant glioma. Brain Res. 2007, 1134, 45–52. [DOI] [PubMed] [Google Scholar]
- Gao J.; Zhang J.; Long Y.; Tian Y.; Lu X. Expression of tankyrase 1 in gastric cancer and its correlation with telomerase activity. Br. J. Cancer 2006, 94, 341–345. [DOI] [PubMed] [Google Scholar]
- Gelmini S.; Quattrone S.; Malentacchi F.; Villari D.; Travaglini F.; Giannarini G.; Della Melina A.; Pazzagli M.; Nicita G.; Selli C.; Orlando C. Tankyrase-1 mRNA expression in bladder cancer and paired urine sediment: preliminary experience. Clin. Chem. Lab. Med. 2007, 45, 862–866. [DOI] [PubMed] [Google Scholar]
- Narwal M.; Venkannagari H.; Lehtiö L. Structural basis of selective inhibition of human tankyrases. J. Med. Chem. 2012, 55, 1360–1367. [DOI] [PubMed] [Google Scholar]
- Busch A.; Johnson K. C.; Stan R. V.; Sanglikar A.; Ahmed Y.; Dmitrovsky E.; Freemantle S. J. Evidence for tankyrases as antineoplastic targets in lung cancer. BMC Cancer 2013, 13, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao R.; Christova T.; Song S.; Angers S.; Yan X.; Attisano L. Inhibition of tankyrases induces axin stabilization and blocks Wnt signalling in breast cancer cells. PLoS One 2012, 7, e48670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers J. R.; Hu L.; Moses A.; Kaboli K.; Papandrea A.; Raymond P. β-catenin/Wnt signaling controls progenitor fate in the developing and regenerating zebrafish retina. Neural Dev. 2012, 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Hao J.; Hong C. C. Cardiac induction of embryonic stem cells by a small molecule inhibitor of Wnt/β-catenin signaling. ACS Chem. Biol. 2011, 6, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronkov A.; Holsworth D. D.; Waaler J.; Wilson S. R.; Ekblad B.; Perdreau-Dahl H.; Dinh H.; Drewes G.; Hopf C.; Morth J. P.; Krauss S. Structural basis and SAR for G007-LK, a lead stage 1,2,4-triazole based specific tankyrase 1/2 inhibitor. J. Med. Chem. 2013, 56, 3012–3023. [DOI] [PubMed] [Google Scholar]
- Chen B.; Dodge M. E.; Tang W.; Lu J.; Ma Z.; Fan C.-W.; Wei S.; Hao W.; Kilgore J.; Williams N. S.; Roth N. G.; Amatruda J. F.; Chen C.; Lum L. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz M. D.; Kirby C. A.; Stams T.; Chin D. N.; Blank J.; Charlat O.; Cheng H.; Cheung A.; Cong F.; Feng Y.; Fortin P. D.; Hood T.; Tyagi V.; Xu M.; Zhang B.; Shao W. [1,2,4]Triazol-3-ylsulfanylmethyl)-3-phenyl-[1,2,4]oxadiazoles: Antagonists of the Wnt pathway that inhibit tankyrases 1 and 2 via novel adenosine pocket binding. J. Med. Chem. 2012, 55, 1127–1136. [DOI] [PubMed] [Google Scholar]
- Bregman H.; Gunaydin H.; Gu Y.; Schneider S.; Wilson C.; DiMauro E. F.; Huang X. Discovery of a class of novel tankyrase inhibitors that bind to both the nicotinamide pocket and the induced pocket. J. Med. Chem. 2013, 56, 1341–1345. [DOI] [PubMed] [Google Scholar]
- Karlberg T.; Markova N.; Johansson I.; Hammarström M.; Schütz P.; Weigelt J.; Schüler H. Structural basis for the interaction between tankyrase-2 and a potent Wnt-signaling inhibitor. J. Med. Chem. 2010, 53, 5352–5355. [DOI] [PubMed] [Google Scholar]
- Nathubhai A.; Patterson R.; Woodman T. J.; Sharp H. E. C.; Chui M. T. Y.; Chung H. H. K.; Lau S. W. S.; Zheng J.; Lloyd M. D.; Thompson A. S.; Threadgill M. D. N3-Alkylation during formation of quinazolin-4-ones from condensation of anthranilamides and orthoamides. Org. Biomol. Chem. 2011, 9, 6089–6099. [DOI] [PubMed] [Google Scholar]
- Pankiewicz K. W. Inhibitors of inosine monophosphate dehydrogenase as potential chemotherapeutic agents. Expert Opin. Ther. Pat. 1999, 9, 55–65. [Google Scholar]
- Hedstrom L. IMP dehydrogenase: Mechanism of action and inhibition. Curr. Med. Chem. 1999, 6, 545–560. [PubMed] [Google Scholar]
- Griffin R. J.; Srinivasan S.; Bowman K.; Calvert A. H.; Curtin N. J.; Newell D. R.; Pemberton L. C.; Golding B. T. Resistance-modifying agents. 5. Synthesis and biological properties of quinazolinone inhibitors of the DNA repair enzyme poly(ADP-ribose) polymerase (PARP). J. Med. Chem. 1999, 41, 5247–5256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.