

Abstract

Herein, we describe the discovery of inhibitors of norepinephrine (NET) and dopamine (DAT) transporters with reduced activity relative to serotonin transporters (SERT). Two compounds, 8b and 21a, along with nomifensine were tested in a rodent receptor occupancy study and demonstrated dose-dependent displacement of radiolabeled NET and DAT ligands. These compounds were efficacious in a rat forced swim assay (model of depression) and also had activity in rat spontaneous locomotion assay.
Keywords: norepinephrine, dopamine, uptake inhibitors, antidepressant
Despite new drugs that have helped many patients manage depressive disorders, there remains a large portion of patients who do not respond to current therapies.1,1b The most frequently prescribed drugs work by blocking uptake of monoamine neurotransmitters such as serotonin (SERT).2 A well-known example of a selective serotonin reuptake inhibitor (SSRI) with clinical efficacy is illustrated by fluoxetine 1 (Figure 1).3 Dual monoamine reuptake inhibitors such as duloxetine 2 have gained clinical acceptance and work by blocking serotonin as well as norepinephrine reuptake (SNRI).4 Triple reuptake inhibitors (TRI) such as amitifadine 4 are currently in clinical studies and demonstrate the ability to block SERT, norepinephrine (NET), and dopamine (DAT) reuptake.5
Figure 1.

Examples of SSRI, SNRI, NDRI, and TRI.
Dual NET/DAT reuptake inhibitors (NDRIs) have also been utilized for the treatment of depression. The only current clinically utilized compound with NDRI activity is buproprion, but the mechanism of action is complex and may involve active metabolites.2 Nomifensine 3 was a clinically useful NDRI but was withdrawn from the market in 1986 due to increased incidence of hemolytic anemia.6−7c
Published reports on nomifensine 3 have hypothesized that this hemolytic anemia might have been associated with a reactive metabolite, specifically formation of a dihydroisoquinolinium ion.8 With this in mind, we sought to identify a novel scaffold with dual NET and DAT inhibition, which would not form a dihydroisoquinolinium ion posing a potential reactive metabolite risk.
On the basis of the known SAR from the literature and in-house examples,9 we hypothesized that the aniline was not necessary for activity, and the fused aromatic ring could be replaced in nomifensine with an cyclic aliphatic ring. Several scaffolds were investigated, but the 2,3,4,7-tetrahydro-1H-azepine proved to be useful in having the desired SAR as well as synthetic handles for further optimization.
Preparation of 2,3,4,7-tetrahydro-1H-azepines is illustrated in Scheme 1. Reaction of acid 5a–j and N-methylprop-2-en-1-amine followed by ring closing metathesis gave the lactams 6a–j.10,10b Reduction of 6a–j with LAH provided N-Me analogues 7a–e and intermediates 7f–j. Demethylation of 7a–j provided compounds 8a–j.
Scheme 1. Preparation of Azepine Analogues.

Reagents and conditions: (a) CH=CHCH2NHMe, 2-chloro-1-methyl pyridinium iodide, CH2Cl2, 25–85%. (b) Grubbs II catalyst,10,10b PhMe, 40 °C, 40–70%. (c) LiAlH4, THF, 20–75%. (d) ACE-Cl, PhMe, reflux, 30–62%.
Enantiomerically pure 2,3,4,7-tetrahydro-1H-azepines were prepared by starting with enantiomerically pure acid to give the (R) stereoisomers 7b and 8b and (S) stereoisomers 7c and 8c.11 Analogues 7a and 8a could be hydrogenated to give the azepanes 9 and 10 (Scheme 2). Alkylation of azepine 8b provided N-alkyl derivates such as 11 and 12.
Scheme 2. Preparation of Azepine and Azepane Analogues.

Reagents and conditions: (a) 7a, H2, Pd/C, HCl, 79%. (b) 8a, PtO2, H2, AcOH. (c) KOH, MeCN/H20, 150 °C, microwave, 75% for 11, 35% for 12.
The preparation of the quaternary substituted 2,3,4,7-tetrahydro-1H-azepines is illustrated in Scheme 3. The hydroxyethyl substituent was selected based on parallel SAR emerging from studies on a piperidine scaffold. The reaction sequence began with 13, which was alkylated with methoxymethyl ether (MOM) protected bromoethanol. A second deprotonation using KOH and tetrabutylammonium iodide (TBAI) as a phase transfer catalyst installed the quaternary center in 14. Reduction of the nitrile with diisobuytlaluminum hydride (DIBAL)-H, followed by hydrolysis, led to an intermediate aldehyde. Reductive amination and protection led to 15, which was subjected to ring-closing metathesis and then deprotected. Chiral supercritical fluid chromatography (SFC) provided enantiomers 16a and 16b. Assignment of absolute configuration was done by X-ray crystallography and confirmed by comparison of measured and computed vibrational circular dichroism (VCD) spectra.12,12b
Scheme 3. Quaternary Substituted Analogues.

Reagents and conditions: (a) BrCH2CH2OMOM, NaH, THF, 42%. (b) CH=CHCH2Br, KOH, TBAI, MeCN, 77%. (c) DIBAL-H, PhMe. (d) CH=CHCH2NH2, NaBH(OAc)3, 1,2-dichloroethane, 37% for steps c and d. (e) Boc2O, CH2Cl2, 78%. (f) Grubbs II catalyst,10b CH2Cl2, reflux, quan. (g) HCl, MeOH, quan., chiral SFC.
Our investigation also examined piperidine analogues with substitution similar to those in the azepine series. Several 2-(3-phenylpiperidin-3-yl)ethanol type derivatives had previously been synthesized in a different project13 and were tested for activity in the NET, DAT, and SERT assays as a direct comparison to the azepines 16a and 16b. From this screening effort, 17a and 17b were identified.
The synthesis of a benzofuran analogue was accomplished in analogous fashion (Scheme 4).14 The enantiomers were separated using chiral SFC to give 21a and 21b and assigned by measured and computed VCD spectra.
Scheme 4. Quaternary Substituted Piperidines.

Reagents and conditions: (a) NaH, Br(CH2)2OMOM. (b) NaH, Br(CH2)3Cl. (c) Ra Ni, NH4OH, NaCl (aq); HCl, MeOH; chiral HPLC.
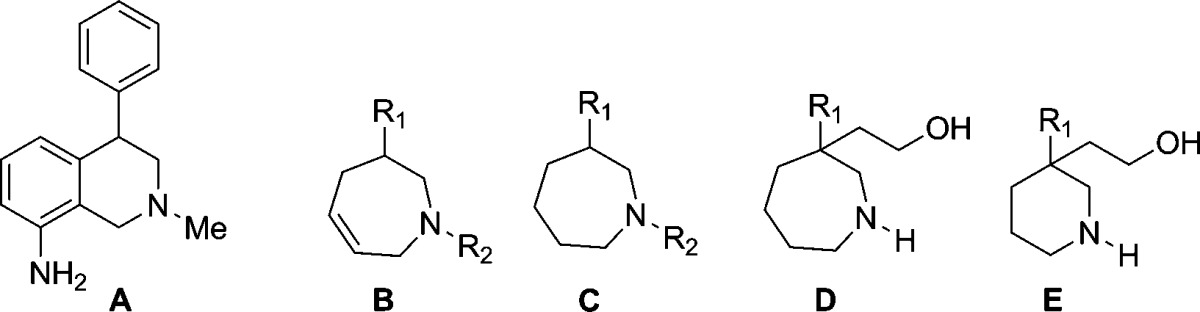
Results of NET, DAT, and SERT testing are reported in Table 1, along with cardiovascular ion channel effects (hERG), hClint, and CYP2D6 profiles. A notable observation in these results is a pronounced difference between the nomifensine stereoisomers [e.g., 3-(S) ≫ 3-(R)], which was confirmed to be consistent with the literature.15 All activity for nomifensine resides in the (S) stereoisomer, although the nomifensine racemate was used clinically. This large dependence on stereochemistry was not observed with the 3,4-dichlorophenyl 2,3,4,7-tetrahydro-1H-azepines such as 7b versus 7c or 8b versus 8c, where only a minor difference was observed. Replacement of the 3,4-dichlorophenyl moiety resulted in a loss of potency at all transporters with the exception of the napthyl analogue 8d (an expected bioisotersic replacement of 3,4-dichlorphenyl). This change led to similar potency at NET and DAT but was more potent at SERT when compared to racemic 3,4-dichlorophenyl 8a. The N–H benzofuran analogue 8j was similar in NET activity to the 3,4-dichlorophenyl analog 8a but showed a slight drop-off in DAT affinity.
Table 1. Summary Results of NET, DAT, and SERT Uptake and Selected in Vitro Properties.
|
Ki (nM) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | core | R1 | R2 | NETa | DATa | SERTa | hERG IC50 (μM)b | hClintc | CYP2D6 IC50 (μM)d |
| 3 | A | 5.0 | 10 | 1400 | >27 | 37 | >20 | ||
| 3 (S) | A | 4.0 | 7.0 | 1200 | NT | NT | NT | ||
| 3 (R) | A | 1200 | 450 | 800 | NT | NT | NT | ||
| 7a | B | 3,4-dichlorophenyl | Me | 57 | 5.0 | 100 | 7.6 | 57 | 10.2 |
| 7b | B | (R)-3,4-dichlorophenyl | Me | 12 | 3.0 | 83 | 10 | 51 | 7.6 |
| 7c | B | (S)-3,4-dichlorophenyl | Me | 54 | 13 | 130 | NT | 86 | 11 |
| 8a | B | 3,4-dichlorophenyl | H | 66 | 6.0 | 220 | 11 | 30 | 5.1 |
| 8b | B | (R)-3,4-dichlorophenyl | H | 17 | 6.0 | 220 | 21 | 33 | 3.9 |
| 8c | B | (S)-3,4-dichlorophenyl | H | 24 | 8.0 | 420 | 13.7 | 50 | 3.1 |
| 8d | B | 2-napthyl | H | 25 | 10 | 63 | 15.4 | 94 | 6.6 |
| 8e | B | 3-Cl-4-Me phenyl | H | 83 | 49 | 600 | >27 | 100 | 5.6 |
| 7d | B | 4-Cl, 3-Me phenyl | Me | 130 | 23 | 270 | 26 | 85 | 18 |
| 8f | B | 4-Cl, 3-Me phenyl | H | 170 | 24 | NT | 26 | NT | NT |
| 8g | B | 3-Cl, 4-F phenyl | H | 130 | 68 | 1500 | 23 | 15 | 6.4 |
| 8h | B | 4-Cl phenyl | H | 250 | 25 | NT | >31 | NT | NT |
| 8i | B | 3-Cl phenyl | H | 430 | 240 | NT | >33 | NT | NT |
| 7e | B | 2-benzofuran | Me | 120 | 60 | 800 | 15 | 97 | 6.7 |
| 8j | B | 2-benzofuran | H | 46 | 23 | 240 | 19 | 83 | 7.8 |
| 9 | C | 3,4-dichlorophenyl | Me | 74 | 8.0 | 260 | 10 | NT | NT |
| 10 | C | 3,4-dichlorophenyl | H | 130 | 11 | 140 | 6.1 | 35 | 2.3 |
| 11 | B | 3,4-dichlorophenyl | -(CH2)2OH | 12 | 37 | 150 | 10 | 52 | >20 |
| 12 | B | 3,4-dichlorophenyl | -(CH2)2OMe | 15 | 9.0 | 540 | 5.3 | 150 | 3.9 |
| 16a | D | (S)-3,4-dichlorophenyl | H | 5.1 | 4.2 | 31 | >33 | <4 | 18 |
| 16b | D | (R)-3,4-dichlorophenyl | H | 49 | 91 | 230 | >33 | NT | NT |
| 17a | E | (S)-3,4-dichlorophenyl | H | 3.3 | 9.3 | 42 | >33 | <4 | >20 |
| 17b | E | (R)-3,4-dichlorophenyl | H | 250 | 140 | 250 | >33 | <4 | >20 |
| 21a | E | (R)-2-benzofuran | H | 4.0 | 6.0 | 230 | >33 | 22 | >20 |
| 21b | E | (S)-2-benzofuran | H | 430 | 400 | NT | NT | NT | NT |
Uptake inhibition measured using HEK cells overexpressed with human recombinant NET, DAT, or SERT receptors with Molecular Devices neurotransmitter kit (inhibition of a fluorescent substrate).17 Results of NET, DAT, and SERT are an average of at least n = 3 with up to 2-fold variability. NT, not tested.
Values determined in CHOK1-hERG cells using electrophysiological measurements.
Human liver microsomal clearance (μL/min/mg).
In vitro inhibition of cytochrome P450 CYP2D6 isoforms.
The activities of racemic saturated azepanes 9 and 10 were similar when compared to the azepine counterparts 7a and 8a. The introduction of groups on the azepine N atom was generally well tolerated for NET and DAT activity. Compounds 11 and 12 illustrate that hydrophilic substituents on the N atom gave the desired NET = DAT > SERT ratio. However, N-alkylation appeared to give rise to more potent activity at hERG (cf. 12 hERG IC50 = 5.3 μM vs 8b = 21 μM) as well as an increase in hClint. Incorporation of a hydroxyethyl substituent (16a, 17a, and 21a) at the 3-position led to an increase in NET but also an increase in SERT. In these cases, a more pronounced difference in stereochemical activity was seen, similar to nomifensine 3.
Incorporation of the hydroxyethyl substituent also improved metabolic stability, decreased hERG, and improved CYP2D6 liability relative to 8b. The benzofuran quaternary substituted piperidine 21a provided the closest match to the nomifensine profile with NET ≅ DAT (Ki = 4.2 and 6.3 nM, respectively) and a large separation at SERT (Ki = 230 nM). Compounds 8b and 21a were also tested for reactive metabolites with GSH under in vitro metabolic activation and did not reveal any adduct formation.
In vivo receptor occupancy was selected as a pharmacodynamic assay to verify that selected ligands could block accumulation of labeled NET/DAT ligands in respective brain regions (Table 2). Test compounds were administered to the rats, followed 30 min later by administration of a radioligand specific for NET or DAT. This assay is described in more detail in the Supporting Information.
Table 2. NET and DAT Receptor Occupancy Studies in Rats.
| compd | NET EC50 (nM)a | DAT EC50 (nM) | NET ED50 (mg/kg)d | DAT ED50 (mg/kg)d | % freee | B/P ratiof |
|---|---|---|---|---|---|---|
| 3 | 3.1 | 31c | 0.05 | 0.7c | 60.5 | 10.1 |
| 8b | 46 | 220b | 0.5 | 2.1c | 7.7 | 4.8 |
| 21a | 5.6 | 33c | 0.17 | 0.8d | 75 | 5.8 |
EC50 = unbound plasma levels equaling 50% occupancy. Utilized [3H]MeNER as NET ligand (sc) and quantified in thalamus.
Utilized [3H]WIN 35,428 as DAT radioligand (sc), quantified in striatum.
Utilized [3H] PE2I (sc) and quantified in striatum.
ED50 = dose to give 50% occupancy at the receptor. The mean value is reported. The 95% confidence interval was typically ± half of the mean value reported. Scatter plots are presented in the Supporting Information to give the reader a more detailed view of the error in the assay.
Percent free plasma concentrations in rat.
Brain to plasma ratio.
Nomifensine was chosen as the benchmark comparator compound, and [3H]-MeNER and [3H]-PE2I were used as the NET and DAT ligands, respectively.16−16c It should also be noted that early in the program, [3H]-WIN35,458 was used as the DAT ligand,16 but it was found that [3H]-PE2I gave smoother curves and a better correlation with our in vitro assays. Compound 8b was tested using [3H]-WIN35,458 but was not repeated with the new ligand. The calculated EC50 in these assays demonstrated a slight disconnect with the human recombinant transporter assays in Table 1, most notably against DAT. This was not unexpected given the differences between rat and human orthologues and the difference between a human recombinant assay and a rat receptor occupancy assay with the inherent errors therein. It is not clear why this discrepancy was more prominent in the DAT measurement. The values of 3.1 and 31 nM observed for nomifensine 3 for NET and DAT, respectively, are consistent with the values reported in a rat synaptosomal uptake assay (NET Ki = 4 nM, DAT Ki = 26 nM).6c This observation led us to believe that the rat receptor occupancy study could be used as a key tool in dose selection for preclinical efficacy testing in rodents. Overall, the receptor occupancy provided rank order in NET and demonstrated potent and dose-dependent receptor occupancy at both receptors at pharmacologically relevant doses used in behavioral assays.
These compounds were then tested in a rat forced-swim assay (FST) for antidepressant effects. Results are shown in Figure 2. Nomifensine 3 demonstrated efficacy at 10, 3, and 1 mg/kg but not at 0.3 mg/kg. It should be noted that doses for the rat forced-swim were selected based on systemic exposure and receptor occupancy, described above. The 2,3,4,7-tetrahydro-1H-azepines 8b demonstrated activity at 10 and 3 mg/kg but not at 1 mg/kg.
Figure 2.

In vivo activity in FST, spontaneous LMA. Top row: Immobility time in FST for compounds 3, 8b, and 21a following dosing (sc) in Sprague–Dawley rats. Middle row: Distance traveled in spontaneous locomotive activity (LMA) in rats as measured by beam breaks. Bottom row: Stereotypic behavior in the locomotive activity assay as measured by number of times the same beam is broken. DMI = desipramine (positive control, dosed 15 mg/kg ip). Data presented as group means ± SEMs. An asterisk indicates statistical significance from vehicle (Veh), using one-way ANOVA analysis.
The quaternary piperidine 21a demonstrated efficacy at 7.3 and 2.4 mg/kg but not at 0.7 mg/kg. The compounds were also examined in a rat spontaneous locomotion assay (LMA) as a behavioral measure of activation of the DAT system. In contrast, selective NET inhibitors have been reported to suppress locomotor activity.18 The compounds were evaluated for both distance traveled and stereotypic behaviors in the last 15 min of the assay. Nomifensine 3 dose dependently induced both locomotor activity and stereotypic behavior with an inverted U-shaped curve. The reductions in LMA could be related to hyperactivation of the dopaminergic system and a shift toward stereotypic behaviors. Alternatively, LMA suppression may be driven by increasing activation of the NET system. Compound 8b exhibited a profile similar to that of nomifensine 3. Although dose-dependent increases in LMA and stereotypy were observed, dose-dependent decreases in LMA and stereotypy were not observed with compound 21a. Perhaps, declinations in these measures might be observed at higher doses. Given the overall precision of the FST, LMA, and stereotypy assays and the challenge in interpreting the behavioral manifestations of activating two major neuromodulatory systems, it can be argued that nomifense 3 and compounds 8b and 21a displayed similar overall behavioral profiles. No severe stereotype such as self-biting was observed with any compound at any dose. Because the objective of the project was to identify an “activating” antidepressant with a DAT uptake inhibition profile, increases in LMA were expected. However, it remains to be determined whether increased activation of the DAT system will afford an adequate therapeutic index.
Plasma levels of 8b were measured at all doses in the FST experiment. On the basis of an assumption that free plasma would equal free brain levels, it was estimated that the free plasma concentration at the lowest effective dose in FST (3 mg/kg) was equivalent to the free concentration that gave between 50 and 70% occupancy at DAT and >90% at NET (based on receptor occupancy studies, see the Supporting Information for more details).
In summary, several potent dual NET/DAT uptake inhibitors have been reported, which have in vivo efficacy similar to nomifensine in a rat model of depression. Further work will need to be done to understand clinical doses and potential dose-limiting side effects such as abuse liability.
Acknowledgments
We thank Amy Hehman, Lynne Neveras, Lisa Leon, Laurie Ross, Donna Sottung, John Zysk, Yan Li, William Potts, Chengwei Fang, and Peter Dorff.
Glossary
Abbreviations
- TBAI
tetrabutylammonium iodide
- DIBAL
diisobuytlaluminum hydride
- MOM
methoxymethyl ether protecting group
- CYP
cytochrome P450 metabolic enzymes
- hERG
human ether a-go-go related gene
- SFC
supercritical fluid chromatography
- ACE
1-chloroethyl chloroformate
Supporting Information Available
Synthetic procedures and assay details. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ AstraZeneca Pharmaceuticals, 35 Gatehouse Drive, Waltham, Massachusetts 02451.
The authors declare no competing financial interest.
Supplementary Material
References
- Nemeroff C. B. Prevalence and Management of Treatment-Resistant Depression. J. Clin. Psychiatry 2007, 68Suppl. 817–25. [PubMed] [Google Scholar]
- Papakostas G. Limitations of Contemporary Antidepressants: Tolerability. J. Clin. Psychiatry 2007, 68, 11–17. [PubMed] [Google Scholar]
- Liu S.; Molino B. F. Recent Developments in Monoamine Reuptake Inhibitors. Annu. Rep. Med. Chem. 2007, 42, 13–26. [Google Scholar]
- Wong D. T.; Perry K. W.; Bymaster F. P. The Discovery of Fluoxetine Hydrochloride (Prozac). Nat. Rev. Drug Discovery 2005, 49764–774. [DOI] [PubMed] [Google Scholar]
- Schatzberg A. F. Efficacy and Tolerability of Duloxetine, a Novel Dual Uptake Inhibitor, in the Treatment of Major Depressive Disorder. J. Clin. Psychiatry 2003, 64Suppl. 1330–37. [PubMed] [Google Scholar]
- Tran P.; Skolnick P.; Czobor P.; Huang N. Y.; Bradshaw M.; McKinney A.; Fava M. Efficacy and Tolerability of the Novel Triple Uptake Inhibitor Amitifadine in the Treatment of Patients with Major Depressive Disorder: A Randomized, Double-Blind, Placebo-Controlled Trial. J. Psychiatric Res. 2012, 46, 692–693. [DOI] [PubMed] [Google Scholar]
- Hunt P.; Kannengiesser M.-H.; Raynaud J.-P. Nomifensine: A New Potent Inhibitor of Dopamine Uptake into Synaptosomes from Rat Brain Corpus Striatum. J. Pharm. Pharmacol. 1974, 265370–371. [DOI] [PubMed] [Google Scholar]
- Tuomisto J. Nomifensine and its Derivatives as Possible Tools for Studying Amine Uptake. Eur. J. Pharmacol. 1977, 422101–106. [DOI] [PubMed] [Google Scholar]
- Nielson J. L.; Lund N. O. Drug Fever due to Nomifensine Treatment in Patients with Endogenous Depression. Int. Pharmacopsychiatry 1981, 16, 66–68. [DOI] [PubMed] [Google Scholar]
- Salama A.; Mueller-Eckhardt C. Two Types of Nomifensine-Induced Immune Haemolytic Anaemias: Drug-Dependant Densitizatioin and/or Autoimmunization. Br. J. Hamaetol. 1986, 64, 613–620. [DOI] [PubMed] [Google Scholar]
- List of Drug Products That Have Been Removed From the Market for Reasons of Safety or Effectiveness. Fed. Regist. 1998, 63 (195), 54082–54089. [PubMed] [Google Scholar]
- Obach S. R.; Dalvie D. K. Metabolism of Nomifensine to a Dihydroisoquinolinium Ion Metabolite by Human Myleoperoxidase, Hemoglobin, Monoamine Oxidase A, and Cythochrome P450 Enzymes. Drug Metab. Dispos. 2006, 3481310–1316. [DOI] [PubMed] [Google Scholar]
- Liu S.; Pechulis A. D.; Beck J. P.; Yang Z.; Vellekoop A. S.; Sweet M. P.; Harms A. E.; Wolf M. A.; Hassler C.; Klos A. M.; Opalks C. J.; Crocker P. J.; Khmelnitsky Y.; Smith M. A.; Molino B. F.. Heterocycle-Fused 4-Phenyl Tetrahydroisoquinolines as Dual NET/DAT Inhibitors, Presented at 243rd ACS National Meeting & Exposition, San Diego, CA, United States, March 25–March 29, 2012; MEDI-372.
- Tuthill P. A.; Seida P. R.; Barker W.; Cassel J. A.; Belanger S.; DeHaven R. N.; Koblish M.; Gottshall S. L.; Little P. J.; Dehaven-Hudkins D. L.; Dolle R. E. Azepinone as a Conformational Constraint in the Design of κ-Opiod Receptor Agonists. Bioorg. Med. Chem. Lett. 2004, 14, 5693–5697. [DOI] [PubMed] [Google Scholar]
- Trnka T; Grubbs M. R. H. The Development of L2X2R=CHR Olefin Metathesis Catalysts: An Organometallic Success Story. Acc. Chem. Res. 2001, 34, 18–29. [DOI] [PubMed] [Google Scholar]
- Sudhakar A. R.; Tang S.. Optically Active Intermediates for the Preparation of Optically Active Substituted Oximes, Hydrazones and Olefins Useful as Neurokinin Antagonists, U.S. Patent 6,265,614, B1 July 24, 2001.
- Nafie L. A.; Dukor R. K. In Applications of Vibrational Optical Activity in the Pharmaceutical Industry; Pivonka D. E., Chalmers J. M., Griffitsh P. R., Eds.; John Wiley and Sons: Chichester, West Sussex, England; pp 129–154 (2–7). [Google Scholar]
- Crawford T. D. Ab initio calculation of molecular chirooptical properties. Theor. Chem. Acc. 2006, 115, 227–245. [Google Scholar]
- Emonds-Alt X.; Gueule P.; Proietto V.; Van Broeck D.. 1-Azoniabicyclo [2.2.1.]heptanes, Method of Preparing Them and Pharmaceutical Compositions in Which they are Present. U.S. Patent 5,679,693, October 21, 1997.
- Chan J. H. T.; Elix J. A.; Ferguson B. A. Anelated Furans. XVII. The Wittig Reaction of Benzofuranones. Aust. J. Chem. 1975, 28, 1097–1111. [Google Scholar]
- Kunstmann R.; Gerhards H.; Kruse H.; Leven M.; Paulus E. F.; Schacht U.; Schmitt K.; Witte P. U. Resolution, Absolute Stereochemistry, and Enantioselective Activity of Nomifensine and Hexahydro-1H-indeno[1,2-b]pyridines. J. Med. Chem. 1987, 30, 798–804. [DOI] [PubMed] [Google Scholar]
- Pogun S.; Scheffel U.; Kuhar M. J. Cocaine Displaces [3H]WIN 35,428 binding to Dopamine Uptake Sites in Vivo More Rapidly than Mazindol or GBR 12909. Eur. J. Pharmacol. 1991, 198, 203–205. [DOI] [PubMed] [Google Scholar]
- [3H]MeNR:Ghose S.; Fujita M.; Morrison P.; Uhl G.; Murphy D. L.; Mozley P. D.; Schou M.; Halldin C.; Innis R. Synapse 2005, 56, 100–104. [DOI] [PubMed] [Google Scholar]
- PE2I:Emond P.; Guilloteau D.; Chalon S. [3H]PE2I: A Radiopharmaceutical for in vivo Exploration of the Dopamine Transporter. CNS Neurosci. Ther. 2008, 14, 47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haunso A.; Buchanan D. J. Pharmacological Characterization of a Fluorescent Uptake Assay for the Noradrenaline Transporter. J. Biomol. Screening 2007, 12, 378–384. [DOI] [PubMed] [Google Scholar]
- Mitchell H. A.; Ahem T. H.; Liles C.; Javors M. A.; Weinshenker D. The Effects of Norepinephrine Transporter Inactivation on Locomotor Activity in Mice. Biol. Psychiatry 2006, 60, 1046–1052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.