

Abstract

This letter describes a series of small molecule inhibitors of IGF-1R with unique time-dependent binding kinetics and slow off-rates. Structure–activity and structure–kinetic relationships were elucidated and guided further optimizations within the series, culminating in compound 2. With an IGF-1R dissociative half-life (t1/2) of >100 h, compound 2 demonstrated significant and extended PD effects in conjunction with tumor growth inhibition in xenograft models at a remarkably low and intermittent dose, which correlated with the observed in vitro slow off-rate properties.
Keywords: Insulin-like growth factor-1 receptor (IGF-1R), time-dependent inhibition, slow off-rate, cancer
Type I insulin-like growth factor receptor (IGF-1R) has been recognized as a key promoter of tumor growth and an activator of cell survival pathways.1−3 Accumulated preclinical evidence strongly suggests the involvement of IGF-1R and its signaling in various stages of multiple types of human cancer including hepatocellular carcinoma (HCC), Ewing’s sarcoma (EwS), multiple myeloma (MM), and nonsmall cell lung carcinoma (NSCLC), where overexpression of IGF-1R and its ligands IGF-I and IGF-II are associated with disease incidence, progression, and poor prognosis.4,5 These preclinical target validation studies around IGF-1R set the foundation for a tremendous undertaking in drug discovery targeting IGF-1R, which has led to various targeted agents in clinical trials.6,7
In recent years, drug-target binary complex binding kinetics have received increasing attention, especially with respect to the dissociation rate of the ligand–receptor complex since an inhibitor with a prolonged target residence time or slower off-rate can result in superior binding affinity.8−11 For a given slow off-rate inhibitor, a transient (<24 h) plasma exposure would, in theory, be sufficient to produce a sustained (24 h) pharmacodynamic (PD) response. In contrast, for a drug lacking slow off-rate properties, sustained drug exposure above a defined minimum effective concentration is typically required in order to maintain sustained PD effects (Figure 1). This difference in such a pharmacokinetic/pharmacodynamic (PK/PD) relationship could further translate into several key advantages for a slow off-rate drug: (1) lower dosage required for sustained target inhibition; (2) less drug-related toxicity as a result of a shorter duration in circulation; (3) larger therapeutic window derived from higher target specificity if the off-rate for the desired drug-target complex is significantly slower than that for off-target complexes;11 (4) in combination studies, potential to avoid drug–drug interactions (DDIs) through a sequential dosing strategy. Various marketed drugs and drug candidates in clinical development, including small molecule kinase inhibitors, have been shown to possess prolonged target residence times or target specific slow off-rate characteristics.8−11 However, the slow off-rate property of reported drug molecules were oftentimes only realized at a mature stage in the drug discovery process and therefore were not subjected to further optimization with respect to binding kinetics.
Figure 1.
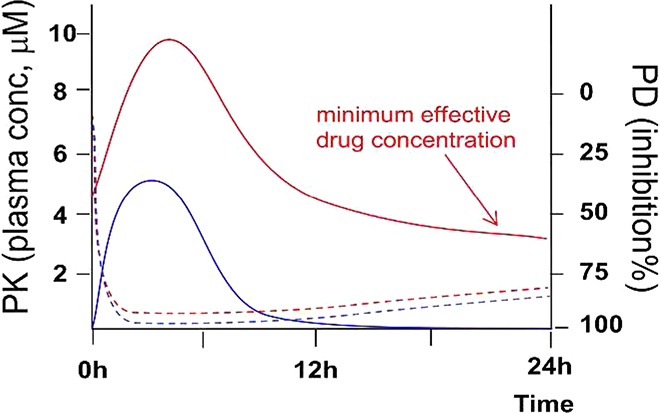
Dissociation kinetics of an inhibitor from its target will have an impact on defining the PK/PD relationship. For a hypothetical slow off-rate drug, a transient (<24 h) plasma exposure (blue solid line) would be sufficient to produce a significant and sustained (24 h) PD response (blue dotted line). However, for a drug with readily reversible dissociation kinetics, a sustained plasma exposure (red solid line) above a minimum effective concentration is typically required to maintain target coverage and therefore a sustained PD response (red dotted line).
Herein, we report a series of compounds with unique time-dependent binding kinetics and slow off-rates against IGF-1R. Specifically, we describe the exploration and optimization of structure–activity relationships (SAR) and structure–kinetic relationships (SKR) leading to the identification of compound 2, a potent, selective, and orally bioavailable IGF-1R inhibitor with slow off-rate and in vivo efficacy at a remarkably low, intermittent dose. To the best of our knowledge, this is the first report describing the systematic exploration and optimization of drug-target dissociation rates based on SKR.
Previously, we disclosed our drug discovery efforts around imidazo[1,5-a]pyrazine12−15 and imidazo[5,1-f][1,2,4]triazine16 derived IGF-1R inhibitors, including the discovery of clinical agent OSI-906 (linsitinib).14 Our IGF-1R inhibitors derived from imidazo[1,5-a]pyrazine scaffold share a 2-phenylquinolinyl moiety, a key pharmacophore deemed critical for both IGF-1R potency and selectivity against other kinases.12−15 During lead optimization efforts involving systematic modification of this quinoline structural unit, we discovered that a methoxy substitution at the C4 position (compound 1, Figure 2) imparted time-dependent inhibition of IGF-1R, as evidenced by the curvature of reaction progress curves from inhibition of in vitro IGF-1R kinase activity by compound 1 as compared to linear curves observed for OSI-906 (Figure 2a,b). We were intrigued by this initial discovery, recognizing that the observation of time-dependent inhibition is often indicative of a slow off-rate. Therefore, we developed a protocol for the determination of the enzyme–compound dissociation rate whereby IGF-1R was incubated with excess compound and then quickly diluted into an assay mixture containing peptide substrate and excess ATP.17 The regain of activity upon dissociation of compound from the enzyme was monitored as a function of time. As shown in Figure 2c, OSI-906 demonstrates readily reversible behavior with full recovery of IGF-1R activity observed within ∼10 min (t1/2 = 0.2 h) of dilution. Reaction kinetics are near-linear and similar to those observed for IGF-1R incubated in the absence of compound (DMSO). In contrast, compound 1, while still exhibiting reversible inhibition of IGF-1R, demonstrated 14-fold slower dissociation kinetics (t1/2 = 2.7 h). Recovery of IGF-1R activity following incubation with compound 1 is biphasic, consistent with our initial observations of time-dependent inhibition. These observations were further confirmed by potency differences in biochemical assays performed with and without preincubation. Compound 1 demonstrated a 10-fold lower IC50 value against IGF-1R following a 24 h preincubation period (7.1 nM (no preincubation) vs 0.7 nM (24 h preincubation)). In contrast, OSI-906 did not demonstrate a shift in IC50 following a preincubation step (12 nM (no preincubation) vs 13 nM (24 h preincubation)).17 Furthermore, the superior biochemical potency of compound 1 over OSI-906 (following preincubation) correlates with superior cellular mechanistic potency (IC50, 3 nM (compound 1) vs 24 nM (OSI-906)).17
Figure 2.
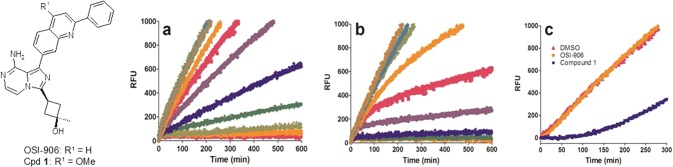
IGF-1R reaction progress and reversibility curves in the presence of OSI-906 and compound 1 in a continuous Omnia assay format. (a) IGF-1R activity is linear with time (R2 > 0.9) in the presence of varying concentrations of OSI-906; (b) IGF-1R progress curves measured in the presence of compound 1 are nonlinear and demonstrate time-dependent inhibition; (c) recovery of IGF-1R activity upon dilution into assay mix (RFU, relative fluorescence units).
Encouraged by the initial observations surrounding compound 1, we followed up with a three tiered plan aimed at identification and in vivo testing of a slow off-rate IGF-1R inhibitor. First, we set out to build a focused library of compounds encompassing a range of IGF-1R off-rates and to broaden the SAR, and more importantly, establish a SKR at the in vitro level. Our second goal was to progress compounds with varying binding kinetics and suitable oral PK profiles to in vivo PD studies to establish a link between in vitro SKR trends and in vivo IGF-1R inhibition. Finally, we sought to advance the most promising compounds to in vivo mouse xenograft efficacy studies. The generation and expansion of the SAR and SKR for this series began with the design of a library of compounds based on varying substituents at the R1, R2, and R3 positions on the imidazo[1,5-a]pyrazine scaffold. Some key analogues from this library are shown in Table 1, where R1 systematically ranges from −OMe to −OPh, R2 is either −H or −F, and R3 is one of three cyclobutyl derived moieties that offered suitable DMPK properties for oral dosing based on previous SAR observations for this series.12−15,17
Table 1. Imidazo[1,5-a]pyrazine Derived IGF-1R Slow off-Rate Inhibitorsa.
| compd | R1 | R2 | inhibitor-IGF-1R half-life (t1/2, h) | IGF-1R cell IC50 (nM)b |
|---|---|---|---|---|
| 1 | OMe | H | 2.7 | 3 |
| 2 | OEt | H | 133 | 4 |
| 3 | OiPr | H | 4.2 | 10 |
| 4 | OPh | H | 5.6 | 23 |
| 5 | OMe | F | 10 | 4 |
| 6 | OEt | F | 995 | 5 |
| 7 | OiPr | F | 43 | 10 |
| 8 | OPh | F | 34 | 8 |
| 9 | OMe | H | 3.3 | 16 |
| 10 | OEt | H | NDc | 24 |
| 11 | OiPr | H | 3.3 | 29 |
| 12 | OPh | H | 14 | 195 |
| 13 | OMe | F | 8.3 | 9 |
| 14 | OEt | F | 743 | 9 |
| 15 | OiPr | F | 11 | 26 |
| 16 | OPh | F | 1.6 | 72 |
See Table 1s in Supporting Information for data for a full list of compounds 1–24.
Cellular mechanistic assay in 3T3/huIGF-1R cell.
Not determined.
As shown in Table 1, these compounds display a broad spectrum of IGF-1R half-lives with t1/2 values ranging from ∼2 to ∼1000 h along with several interesting trends in SKR. For example, all compounds with a substituent other than hydrogen at R1 had some degree of time-dependent behavior, with R1 = OEt analogues having the most extended off-rates. Furthermore, incorporating F for H at R2 provided analogues, which generally trended toward more extended off-rates. However, R3 moieties do not appear to have a significant impact on the dissociation rate. Overall, these SKR trends are generally in agreement with the modeled binding mode of these compounds, where the R1/R2 substituted 2-phenylquinolinyl moiety is a main potency driver, making key interactions with a hydrophobic pocket within IGF-1R, whereas the R3 moiety extends toward a solvent exposed region of the protein.12−15 In terms of their IGF-1R cellular mechanistic potencies, although all of the R1 substitutions were generally tolerated, −OMe and −OEt were clearly preferred substitutions and afforded equivalent IGF-1R potencies in most cases, while −OPh and −OiPr substituted compounds tended to be less potent. Analogues with other types of R1 substitutions such as small alkyl groups were also well tolerated for IGF-1R potency (data not shown).
With potent compounds possessing a diverse array of off-rates in hand, we were particularly interested in evaluating, in the in vivo setting (xenograft models), a matched pair that had similar cellular potencies and PK profiles but significantly different off-rates. Such a matched pair would enable the evaluation of target inhibition (PD) and in vivo efficacy with respect to in vitro biochemical off-rates. Compounds 1 and 2 emerged as an ideal matched pair based upon this criteria. Both compounds were orally bioavailable and had similar overall mouse PK profiles (Table 2). At a dose of 5 mg/kg, compound 1 was rapidly absorbed (Tmax = 0.5 h) with a plasmic Cmax of 3.2 μM. The plasmic drug concentration at 8 h (C8h) was slightly below its cellular IC50 in the presence of 90% mouse plasma protein (cell + mpp IC50) and by 24 h (C24h) was below the limit of detection (0.03 μM). Compound 2 at a dose of 2.5 mg/kg displayed a similar overall PK profile to that of compound 1, which included being rapidly absorbed (Tmax = 0.5 h), a plasmic Cmax of 2.2 μM, a plasmic C8h just below its cell + mpp IC50, and essentially no drug in circulation by 24 h.
Table 2. Mouse PK Profile and Cellular Potency with Mouse Plasma Protein of Matched Pair Compounds 1 and 2.
| iv dose |
po dose |
|||||||
|---|---|---|---|---|---|---|---|---|
| compd | Cl (mL/min/kg) | Vss (L/kg) | Cmax (μM) | Tmax (h) | C8h (μM) | C24h (μM) | F% | cell + mPP IC50 (μM)c |
| 1a | 17 | 2.4 | 3.2 | 0.5 | 0.28 | <0.03 | 74 | 0.32 |
| 2b | 5 | 1.5 | 2.2 | 0.5 | 0.32 | <0.03 | 63 | 0.44 |
A 0.5 mg/kg iv dose and a 5 mg/kg po dose.
A 5 mg/kg iv dose and a 2.5 mg/kg po dose.
Cellular mechanistic IC50 measured in the presence of 90% mouse plasma protein.
A single dose PD study for compound 2 was carried out in a human colon cancer (GEO) xenograft model, which harbors an IGF-1R/IGF-II autocrine loop.12 We were excited to see that at an oral dose of 2 mg/kg of compound 2 showed a significant and sustained PD response, inhibiting the phosphorylation of both IGF-1R and IR18 (∼80%) for up to 48 h (Figure 3a). This dramatic PD response extended well beyond the availability of drug in plasma and is in line with the slow off-rate observed in vitro. Encouraged by this result, compound 2 was progressed to a 14-day efficacy study in the same model. Tumor growth inhibition (TGI)17 of 66% was observed at a remarkably low, intermittent, and well-tolerated dose of 2 mg/kg given orally once every 7 days (Figure 3b). For comparison, compound 1 was progressed to a GEO xenograft study where 64% TGI was observed at a dose of 5 mg/kg QD (Figure 3c). Unlike compound 2, the intermittent dose for compound 1 did not show significant TGI (<50%, data not shown), and daily dosing (QD) was required for efficacy comparable to compound 2 dosed on the intermittent schedule. Taken together, these data suggest that for these IGF-1R inhibitors, SKR can be optimized to afford compounds with varying off-rates. Furthermore, extended off-rates, exemplified by compound 2, can translate to extended target coverage and in vivo efficacy.
Figure 3.
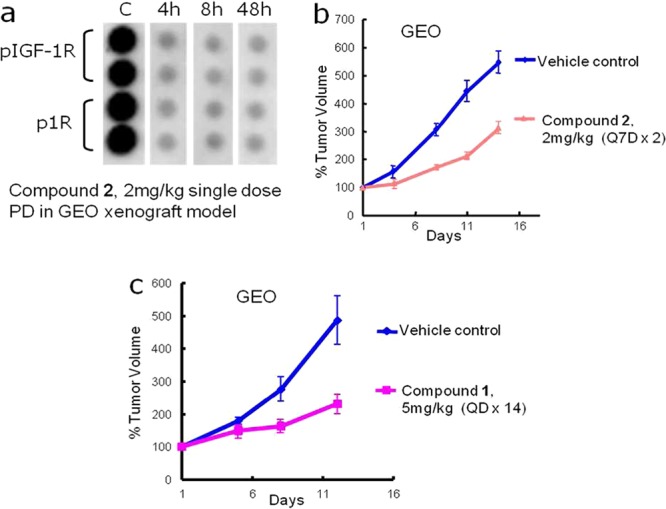
In vivo PD and TGI studies in GEO tumor xenografts. (a) Compound 2: single dose PD study at 2 mg/kg dose, sustained inhibition of pIGF-1R and pIR was observed for 48 h. (b) Compound 2: 66% TGI observed at 2 mg/kg every 7 days (Q7D × 2) dose. (c) Compound 1: 64% TGI observed at 5 mg/kg every day (QD 1–14). Plotted data are mean tumor volumes expressed as a percentage of initial volume ± SE.
The kinase selectivity of compound 2 was evaluated in-house by screening against a panel of 192 kinases using a Caliper EZ Reader mobility shift assay. Compound 2 achieved >90% inhibition of IGF-1R and IR without significant inhibition (<50%) of any other kinases in the panel; thus, it is a highly selective inhibitor. As part of the pharmaceutical development assessment, compound 2 was also progressed through a series of preclinical ADMET assays. As shown in Table 3s (see Supporting Information), it demonstrated robust microsomal stability in vitro toward multiple species suggesting an expected low first pass metabolism (mouse microsomal extraction ratios (ER) of 0.28 translates to low clearance of 5 mL/min/kg in vivo). Compound 2 has good aqueous solubility especially at a lower pH, is highly permeable, and has a clean CYP450 inhibition profile against 5 major isoforms. The ligand-lipophilic efficiency (LLE)19 for compound 2, based on a preincubation biochemical IC50 value of 0.6 nM, is favorable at 5.13. These promising physicochemical properties, taken together with encouraging pharmacological results both in vitro and in vivo, suggest overall favorable drug-like properties for compound 2.
In summary, a series of small molecule IGF-1R inhibitors with unique time-dependent inhibition and slow dissociation kinetics were discovered and developed. Both SAR and SKR were established through a systematic medicinal chemistry effort. Representative compounds were progressed into in vivo PD/efficacy studies. Most notably, compound 2, a highly potent, selective, and orally bioavailable IGF-1R slow off-rate inhibitor, produced significant tumor growth inhibition at a remarkably low, intermittent dose. Analysis of in vitro and in vivo data generated from compounds 1 and 2 demonstrate that drug-receptor off-rates can dramatically influence the overall profile of a drug molecule. These studies underscore the importance of incorporating characterization of binding kinetics into the drug discovery process.
Acknowledgments
We gratefully acknowledge Dr. Yingjie Li and Ms. Viorica M. Lazarescu for analytical support; Mr. Paul Maresca, Ms. Brianna Tokar, Mr. Pete Meyn, Mr. Roy Turton, and the Leads Discovery Group for conducting in vitro ADMET studies; Ms. Kristen Chang for supporting cellular assays; the process chemistry group for providing key intermediates for synthesis of compounds described in this manuscript; Drs. Maryland Franklin, Elizabeth Rizzo, and Qun-Sheng Ji for their overarching contributions to OSI’s IGF-1R programs.
Supporting Information Available
Table 1s, Table 3s, synthetic procedure, analytical data for compound 1 and 2, and procedures for in vitro and in vivo assays. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Kaiser U.; Schardt C.; Brandscheidt D.; Wollmer E.; Havemann K. Expression of insulin-like growth factor receptors I and II in normal human lung and in lung cancer. J. Cancer Res. Clin. Oncol. 1993, 119, 665–668. [DOI] [PubMed] [Google Scholar]
- Kondo M.; Suzuki H.; Ueda R.; Osada H.; Takagi K.; Takahashi T.; Takahashi T. Frequent loss of imprinting of the H19 gene is often associated with its overexpression in human lung cancers. Oncogene 1995, 10, 1193–1198. [PubMed] [Google Scholar]
- Rubin R.; Baserga R. Insulin-like growth factor-I receptor. Its role in cell proliferation, apoptosis, and tumorigenicity. Lab. Invest. 1995, 73, 311–331. [PubMed] [Google Scholar]
- LeRoith D.; Roberts C. T. Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003, 195, 127–137. [DOI] [PubMed] [Google Scholar]
- Ma J.; Giovannucci E.; Pollak M.; Leavitt A.; Tao Y.; Gaziano J. M.; Stampfer M. J. A prospective study of plasma C-peptide and colorectal cancer risk in men. J. Natl. Cancer Inst. 2004, 96, 546–553. [DOI] [PubMed] [Google Scholar]
- Jin M.; Wang J.; Buck L.; Mulvihill M. Small molecule ATP-competitive dual IGF-1R and IR inhibitors: Structural insights, chemical diversity and molecular evolution. Future Med. Chem. 2012, 4, 315–328. [DOI] [PubMed] [Google Scholar]
- Li R.; Pourpak A.; Morris S. W. Inhibition of the insulin-like growth factor-1 receptor (IGF1R) tyrosine kinase as a novel cancer therapy approach. J. Med. Chem. 2009, 52, 4981–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Tonge P. J. Drug-target residence time: critical information for lead optimization. Curr. Opin. Chem. Biol. 2010, 14, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Monsma F. The importance of drug-target residence time. Curr. Opin. Drug Discovery Dev. 2009, 12, 488–496. [PubMed] [Google Scholar]
- Copeland R. A. Conformational adaptation in drug-target interactions and residence time. Future Med. Chem. 2011, 3, 1491–1501. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discovery 2006, 5, 730–739. [DOI] [PubMed] [Google Scholar]
- Ji Q.; Mulvihill M.; Rosenfeld-Franklin M.; Cooke A.; Feng L.; Mak G.; O’Connor M.; Yao Y.; Pirritt C.; Buck E.; Eyzaguirre A.; Arnold L.; Gibson N.; Pachter J. A novel, potent, and selective insulin-like growth factor-I receptor kinase inhibitor blocks insulin-like growth factor-I receptor signaling in vitro and inhibits insulin-like growth factor-I receptor dependent tumor growth in vivo. Mol. Cancer Ther. 2007, 6, 2158–2167. [DOI] [PubMed] [Google Scholar]
- Mulvihill M.; Ji Q.; Coate H.; Cooke A.; Dong H.; Feng L.; Foreman K.; Rosenfeld-Franklin M.; Honda A.; Mak G.; Mulvihill K.; Nigro A.; O’Connor M.; Pirrit C.; Steinig A.; Siu K.; Stolz K.; Sun Y.; Tavares P.; Yao Y.; Gibson N. Novel 2-phenylquinolin-7-yl-derived imidazo[1,5-a]pyrazines as potent insulin-like growth factor-I receptor (IGF-IR) inhibitors. Bioorg. Med. Chem. 2008, 16, 1359–1375. [DOI] [PubMed] [Google Scholar]
- Mulvihill M.; Cooke A.; Rosenfeld-Franklin M.; Buck E.; Foreman K.; Landfair D.; O’Connor M.; Pirrit C.; Sun Y.; Yao Y.; Arnold L.; Gibson N.; Ji Q. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [DOI] [PubMed] [Google Scholar]
- Jin M.; Kleinberg A.; Cooke A.; Gokhale P. C.; Foreman K.; Dong H.; Siu K. W.; Bittner M. A.; Mulvihill K. M.; Yao Y.; Landfair D.; O’Connor M.; Mak G.; Pachter J. A.; Wild R.; Rosenfeld-Franklin M.; Ji Q.; Mulvihill M. J. Potent and selective cyclohexyl-derived imidazopyrazine insulin-like growth factor 1 receptor inhibitors with in vivo efficacy. Bioorg. Med. Chem. Lett. 2011, 21, 1176–1180. [DOI] [PubMed] [Google Scholar]
- Jin M.; Gokhale P.; Cooke A.; Foreman K.; Buck E.; May E.; Feng L.; Bittner M.; Kadalbajoo M.; Landfair D.; Siu K.; Stolz K.; Werner D.; Laufer R.; Li A.; Dong H.; Steinig A.; Kleinberg A.; Yao Y.; Pachter J.; Wild R.; Mulvihill M. J. Discovery of an orally efficacious imidazo[5,1-f][1,2,4]triazine dual inhibitor of IGF-1R and IR. ACS Med. Chem. Lett. 2010, 1, 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information for detailed assay protocols and synthetic procedures.
- Compounds described in this letter share similar potency against IGF-1R and IR, due to high sequence homology between two proteins (overall protein sequence homology, 84%; ATP binding pocket sequence homology, 100%). These compounds also demonstrated very similar SKR trends for IGF-1R and IR (data not shown).
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.