

Abstract
The human neuraminidase enzymes (hNEU) play important roles in human physiology and pathology. The lack of potent and selective inhibitors toward these enzymes has limited our understanding of their function and the development of therapeutic applications. Here we report the evaluation of a panel of compounds against the four human neuraminidase isoenzymes. Among the compounds tested, we identified the first selective, nanomolar inhibitors of the human neuraminidase 4 enzyme (NEU4). The most potent NEU4 inhibitor (5-acetamido-9-[4-hydroxymethyl[1,2,3]triazol-1-yl]-2,3,5,9-tetradeoxy-d-glycero-d-galacto-2-nonulopyranosonic acid) was found to have an inhibitory constant (Ki) of 30 ± 19 nM and was 500-fold selective for its target over the other hNEU isoenzymes tested in vitro (NEU1, NEU2, and NEU3). This is the first report of any inhibitor of hNEU with nanomolar potency, and this confirms that the 2,3-didehydro-2-deoxy-N-acetylneuraminic acid (DANA) scaffold can be exploited to develop new, potent, and selective inhibitors that target this important family of human enzymes.
Keywords: Neuraminidase, sialidase, NEU1, NEU2, NEU3, NEU4, inhibitors, sialic acid, glycosyl hydrolase
Although significant effort has been devoted to the development of potent inhibitors for the viral neuraminidase enzymes (vNEU),1 the family of human neuraminidase enzymes (NEU1–NEU4) have not enjoyed as much attention. One reason for this disparity may be the lack of specific pathologies associated with hNEU. Genetic deficiency of hNEU1 leads to systemic disorders, sialidosis, and galactosialidosis caused by lysosomal storage of sialoglycoconjugates.2 While the hNEU family has been explored for nearly two decades, evidence for their specific biochemical roles has emerged more slowly.3−5 In particular, specific isoenzymes of hNEU have been recently reported to play important roles in inflammation,6,7 platelet clearance,8 insulin signaling,9 and cancer,10 thus highlighting the need for new tools to explore their role in human physiology.5 Thus, chemical inhibitors for this class of enzymes are needed to explore a range of pathologies.11 Importantly, these new chemical tools must provide selective targeting of the enzymes in order to avoid potentially toxic side effects and to discriminate between hNEU isoenzymes.
There are few reports of selective inhibitors against hNEU, and the most potent inhibitors currently known are of mid- to low-micromolar activity. Magesh et al. reported compounds selective for NEU1 (IC50 ∼ 10 μM).12 Inhibitors which target hNEU over bacterial NEU have been reported.13,14 Potent inhibitors of vNEU have only moderate activity against hNEU, with zanamivir being the most potent of these against NEU2 (IC50 ∼ 16 μM).15 Our group has previously examined the activity of oseltamivir derivatives that show mild selectivity for NEU3 over NEU4.16 To address this lack of selective inhibitors, we recently tested a series of C4, C7-modified DANA-analogs designed to target specific isoenzymes of hNEU. This study identified compounds with 12- to 40-fold selectivity against NEU2 and NEU3, respectively.17 However, all these previous reports of inhibitors for hNEU have failed to identify compounds with sufficient potency or selectivity to be used in vivo.
In a previous report, we designed a series of C9-modified derivatives of DANA and tested their activity against NEU3.18 After our recent development of assays for the remaining three hNEU isoenzymes,17 we retested members of the original C9-compound series. These experiments led to the identification of compounds with unprecedented potency and selectivity for the NEU4 isoenzyme.
Our library of C9-modified DANA derivatives exploited Cu catalyzed azide–alkyne cycloaddition (CuAAC) between a panel of alkynes with C9 or N5-azido forms of DANA. Since initial testing of this library identified inhibitors with improved activity against NEU3 relative to the parent DANA structure, we hypothesized that some of these compounds could have potency against other isoenzymes. Therefore, we tested a subset of the original library against the four hNEU isoenzymes (Figure 1).
Figure 1.
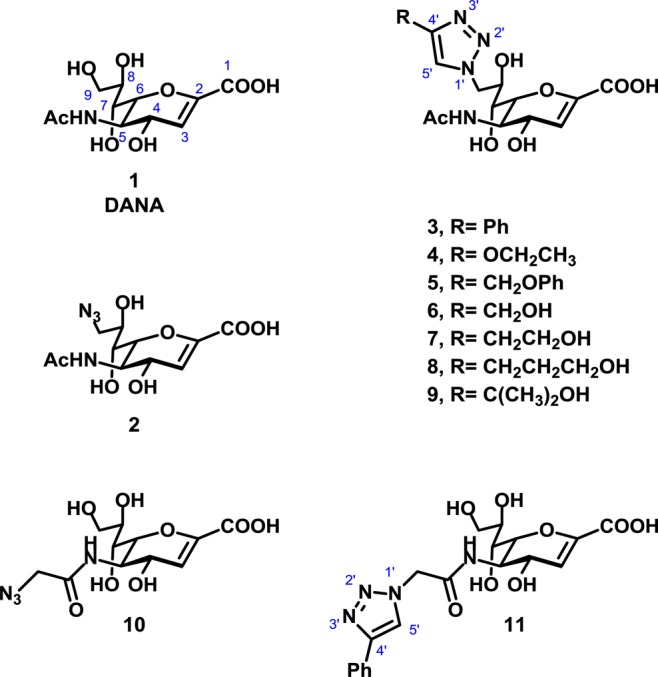
Structures of the compounds used in this study (1–11).
The identity of samples used in this study was confirmed by high resolution mass spectrometry, and HPLC was used to confirm that all samples were >95% purity (see Supporting Information). We then proceeded to test compounds for their ability to inhibit NEU1, NEU2, NEU3, and NEU4 enzymes.18
The results of enzyme inhibition assays are summarized in Table 1. We confirmed that the parent compound, DANA, had broad activity against all four isoenzymes (7–90 μM inhibition).12,15,17 Modification of DANA to the C9-azido derivative (2) resulted in decreased activity against all isoenzymes tested, with the greatest loss in potency observed against NEU1. The C4′-phenyl-C9-triazole derivative of DANA (3) had similar potency for NEU3 (4.6 ± 0.2 μM) and NEU4 (2.3 ± 0.2 μM) but little activity against NEU1 and NEU2 (>200 μM). The addition of a hydrogen bond acceptor in the C9-triazole moiety (4) resulted in moderate selectivity for NEU4 (3.6 ± 0.3 μM) over other isoenzymes (25-fold selectivity). The C4′-methylphenoxy-C9-triazole analog of DANA (5) was over 200-fold selective for NEU3 (5.5 ± 0.6 μM) and NEU4 (1.7 ± 0.2 μM) isoenzymes. Truncation of the C4′-side chain to a hydroxymethyl group (6) provided the most potent compound in our study. Compound 6 was found to have nanomolar potency for NEU4 (0.16 ± 0.01 μM) yet maintained only micromolar activity against other isoenzymes (500-fold). A similar, though not as drastic, trend was observed for the C4′-hydroxyethyl derivative 7 (NEU4, 0.81 ± 0.04 μM). Further extension of the C4′ side chain to the hydroxypropyl derivative, 8, decreased potency for NEU4 (2.1 ± 0.1 μM), whereas activity against NEU3 remained only moderate (39 ± 1 μM). The branched C4′-side chain of compound 9 provided a minor improvement in selectivity for NEU4; however, potency against the enzyme dropped by approximately 2-fold relative to that of compound 8 (4.0 ± 0.3 μM).
Table 1. Inhibition of Neuraminidase Isoenzymes.
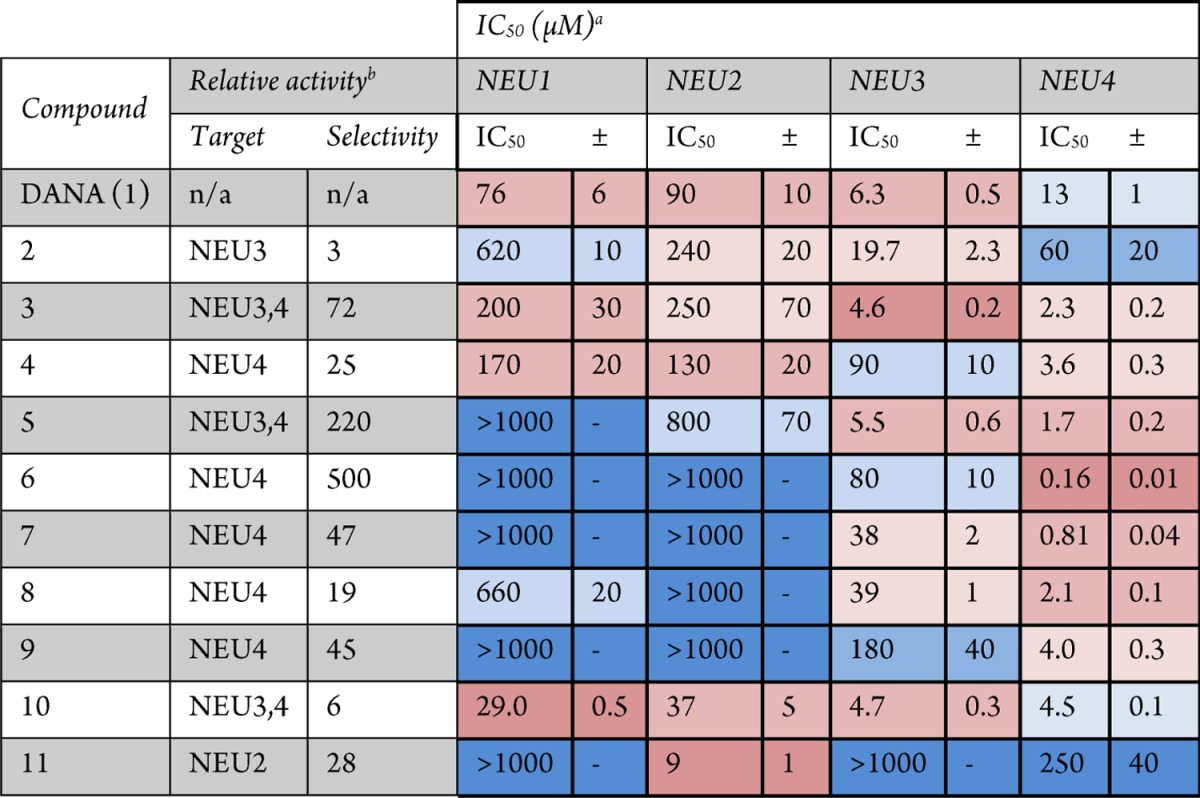
Coloring of potency values is by relative ranking within each isoenzyme. Darker shades of red indicate higher potency compounds with activity below 500 μM, or which are ranked 1–7 among compounds tested. Darker shades of blue indicate weaker potency, with activities typically above 500 μM or which are ranked 8–11 among compounds tested.
Relative activity was determined by dividing the potency of the compound for its next weakest target by that of its primary target. For cases where multiple isoenzymes are listed as the target, the average of these was used.
The N5-azidoacetate derivative of DANA (10) had previously been reported to have improved activity against NEU318 and NEU2.14 In this study we tested the compound against all four hNEU isoenzymes and found that it was most potent against NEU3 (4.7 ± 0.3 μM) and NEU4 (4.5 ± 0.1 μM) and had approximately 6-fold lower activity against NEU1 (29 ± 0.5 μM) and NEU2 (37 ± 5 μM). Testing of a DANA analog which included a triazole group at the N5-position (11) revealed that only the NEU2 isoenzyme could tolerate this bulky side chain (9 ± 1 μM). Importantly, compound 11 was approximately 30-fold selective for NEU2 over the other isoenzymes and was the most potent compound for NEU2 among those tested here.
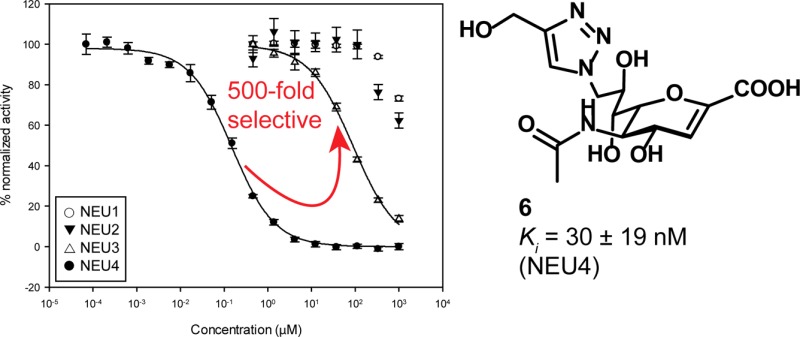
The remarkable potency of compound 6 against NEU4 led us to examine its activity in more detail. An overlay of the inhibition curves for compound 6 against hNEU showed a clear separation between its activity for NEU4 and all other isoenzymes, establishing that the selectivity of the inhibitor was at least 500-fold over its next most active target (NEU3; Figure 2). We then determined the inhibition constant (Ki) for compounds 6 and 7 against NEU4 (Supporting Information). These experiments found that compounds 6 and 7 had Ki values against NEU4 of 30 ± 19 nM and 60 ± 16 nM, respectively, making them the most potent and selective inhibitors reported for any hNEU enzyme. In order to understand the observed selectivity of these compounds in more detail, we developed a molecular model of the interaction of compound 6 with its target, NEU4. No crystal structure data has been reported for NEU4 to date, so we used homology modeling with NEU2 as a template (see Experimental Procedures).19 After docking of compound 6 to the active site of the NEU4 homology model and subsequent molecular dynamics, we obtained a binding model (Figure 3) which maintained most of the expected contacts with the key features of the DANA core. Interestingly, we observed that the 4′-hydroxymethyl group of compound 6 was able to engage in H-bond contacts with the carbonyl groups of S243 and W274 and the side chain of R242 (Figure 3b). Thus, our model suggests that the precise placement of the 4′-hydroxymethyl group is responsible for the remarkable activity of this compound against NEU4. This conclusion is also consistent with the activity of compounds 7 and 8, which both show successive drops in potency with the homologation of methylene groups at the 4′ position. Our model of NEU4 also suggests that the lack of specificity for other isoenzymes, such as NEU2, is the result of differences in the glycerol side chain binding pocket. An alignment of the NEU4 model to NEU2 finds a large conformational change between a loop of the enzyme that forms half of the binding pocket (see Supporting Information). We attribute the difference in activity of compound 6 to this conformational change.
Figure 2.

Potency and selectivity of compound 6 against hNEU. The potency of compound 6 was determined using the 4-MU-NANA assay. IC50 curves of the compound with NEU1, NEU2, NEU3, and NEU4 are overlaid. The IC50 against NEU4 was 160 ± 10 nM, with a selectivity of at least 500-fold over the other three isoenzymes.
Figure 3.
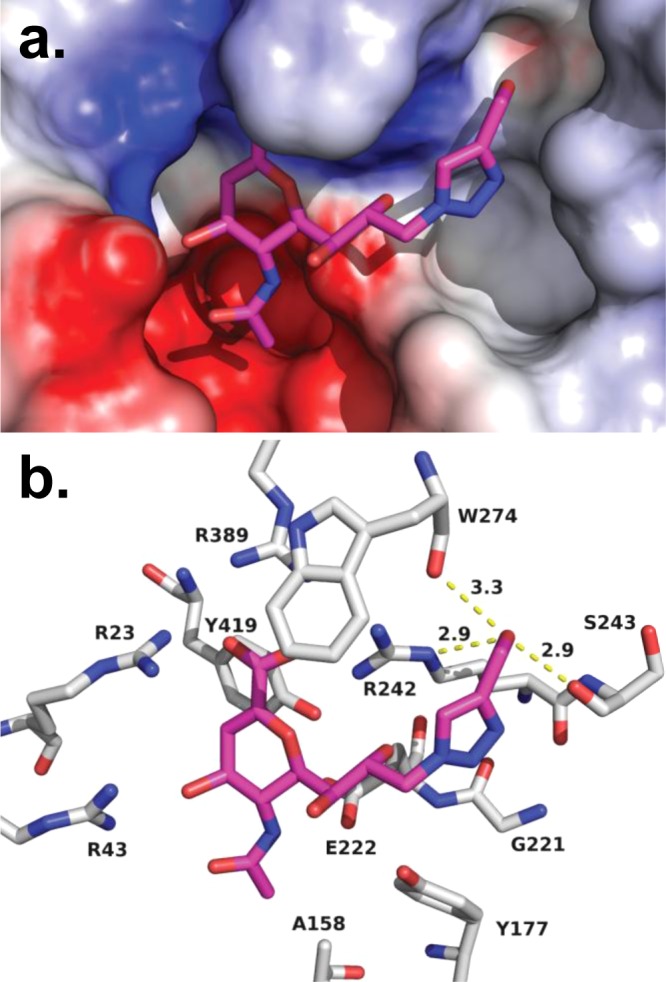
Molecular model of compound 6 in the active site of NEU4. Using our homology model of NEU4, compound 6 was docked to the active site and then subjected to molecular dynamics (10 ns). (a) An electrostatic surface representation of the active site is shown with compound 6 bound. (b) The general binding mode observed for DANA derivatives observed for NEU2 was preserved in our model, including contacts with the arginine triad (R23, R389, and R242). H-bond contacts are maintained between O4 and R43, as well as the glycerol side chain O8 with R242. The C4′–CH2OH has multiple H-bond contacts which include the backbone carbonyls of S243 (2.9 Å) and W274 (3.3 Å) and the Nε of R242 (2.9 Å).
Our results suggested that 6 was a potent competitive inhibitor of NEU4; however, the above assays exploit the fluorogenic substrate, 4MU-NANA, which is not a native substrate of the enzyme. To confirm that this class of inhibitors could, indeed, inhibit the cleavage of native NEU4 substrates, we adapted a reported assay which detects the generation of free sialic acid.20 We tested our most potent NEU4 inhibitor, compound 6, for its ability to inhibit enzyme cleavage of GM3, a known glycolipid substrate of NEU4 (Figure 4).21 We found that the IC50 of 6 with GM3 as a substrate was in the high nanomolar range (740 ± 70 nM) and was 350-fold more potent than DANA. These data confirmed that compound 6 is a potent inhibitor of NEU4 cleavage of native glycolipid substrates.
Figure 4.

Compound 6 inhibits NEU4 cleavage of a glycolipid substrate, GM3. An IC50 curve demonstrating the inhibition of glycolipid hydrolysis catalyzed by NEU4 with compound 6.20 The determined IC50 for 6 was 0.74 ± 0.07 μM, and that for DANA was 260 ± 40 μM, demonstrating an approximately 350-fold difference in activity. Assays were performed with four replicates for each point; error bars indicate the standard deviation.
In this report, we have identified inhibitors which selectively target isoenzymes of hNEU. Our most potent compounds, 6 and 7, are active against NEU4, with nanomolar Ki values. Several of the compounds are also specific against NEU4, with selectivities that ranged from 50-fold (9 and 7) to 500-fold (6). These inhibitors were also demonstrated to act as nanomolar inhibitors of NEU4 processing of the ganglioside substrate, GM3. We also observed that DANA analogs containing a bulky N5 group resulted in selective inhibition of NEU2 (28-fold, 11). These results provide evidence that minor modifications of the DANA scaffold can generate potent and selective inhibitors of hNEU. Additionally, the range of activity and selectivity found among the panel of C9-modified compounds (2–9) supports our hypothesis that the binding sites for the glycerol side chain of Neu5Ac are diverse among hNEU.16,18 NEU4 was identified following the sequencing of the human genome, almost two decades later than other hNEU, but multiple studies have already implicated it in vital functions, such as processing of brain gangliosides,22 neuronal development,23 and cancer.24 The development of specific and potent small molecule inhibitors of this enzyme will therefore provide a vital tool for future studies of pathways controlled by NEU4 and reveal new avenues of therapeutic intervention.
Experimental Procedures
Characterization of Inhibitors
Inhibitors used in this study were prepared as described previously.18 Compound purity was determined by HPLC and confirmed to be ≥95% for all compounds tested. Compound identity was confirmed by high resolution mass spectrometry (see Supporting Information for details).
Inhibition Assays
NEU3 and NEU2 were expressed in E. coli as N-terminal MBP fusion proteins and purified as described previously.25 NEU4 was expressed as a GST fusion protein and purified as described.16 NEU1 was purified as previously described.26 Assays were conducted in 0.1 M sodium acetate buffer at the enzyme optimum pH (4.5 for NEU1, NEU3, and NEU4; 5.5 for NEU2), using a similar amount of enzymatic activity for all four proteins, as determined by assay with 4MU-NANA. Inhibitors were subjected to 3-fold serial dilutions starting from a final concentration of 1 mM. Dilutions were performed in reaction buffer (20 μL). The mixture was then incubated for 15 min at 37 °C. Fluorogenic substrate (4MU-NANA, 50 μM final concentration) was added to the reaction buffer (20 μL) and incubated at 37 °C for 30 min. The reaction was quenched with 200 μL of 0.2 M sodium glycinate buffer pH = 10.7, and enzyme activity was determined by measuring fluorescence (λex = 365 nm; λem = 445 nm) in a 384 well plate using a plate reader (Molecular Devices, Sunnyvale, CA). Assays were performed with four replicates for each point; error bars indicate the standard deviation. Reported IC50 values were determined by nonlinear regression using SigmaPlot 12. For curves which showed less than a 50% decrease in signal, fits were conducted using the maximum inhibition values found for DANA.
To test the potency of inhibitors against a glycolipid substrate, we adapted a known assay for the detection of free sialic acid.20 The assay was conducted in 0.1 M sodium acetate buffer (pH 4.5) with NEU4. The reaction mixture was incubated for 30 min at 37 °C. Substrate (GM3, 500 μM final concentration) was added to the reaction mixture and incubated at 37 °C for 1 h. The reaction was quenched with 200 μL of 0.2 M sodium borate buffer (pH 9.5). A fluorescent product was developed by the addition of malononitrile (40 μL of 0.8% solution) and heating at 80 °C for 20 min. Enzyme activity was determined by measuring fluorescence (λex = 357 nm excitation; λem = 443 nm) in a 384 well plate. Reported IC50 values were determined by nonlinear regression.
Ki Determinations
Solutions of 4MU-NANA in sodium acetate buffer (0.1 M) at the optimum pH of the target enzyme were prepared with concentrations of 20, 40, 60, 80, and 100 μM. Each substrate solution (25 μL) was mixed with an equal volume of a solution containing serial concentrations of the inhibitor and the target enzyme. The inhibitor concentrations were selected as a range which included the determined IC50 value. The reaction mixture (50 μL) was transferred to a 384-well plate. The rate of the product formation at 37 °C was followed by measuring the fluorescence (λex = 365 nm; λem = 445 nm) every 30 s for 60 min using a plate reader (Molecular Devices, Sunnyvale, CA). Pseudo-first-order rates were determined using the initial linear part of the resulting curves. The double reciprocal plot (1/rate versus 1/[S]) for each inhibitor concentration was used to determine a slope, and a plot of the slopes versus inhibitor concentration was fit to determine Ki.
Molecular Modeling
The NEU2 structure reported by Chavas et al. was used to develop a homology model of NEU4 (PDB ID: 1VCU).19 NEU4 and NEU2 sequences (accession numbers: Q8WWR8 and Q9Y3R4, respectively) were aligned using ClustalW227 and found to have 43% homology. Although the extended sequence of NEU4L (residues 287–374) did not align with any parts of NEU2, the full sequence was used to generate a preliminary homology model of NEU4 using SWISS-MODEL.28 The sequence between residues 287 and 374 remained as an unorganized loop in the initial model. The model was first minimized and then DANA was docked to the active site, providing the initial structure for molecular dynamics calculations using MacroModel 9.9. The protein–ligand complex was equilibrated for 100 ps at 300 K, followed by 10 ns of dynamics at constant temperature with 1.5 fs time steps (see Supporting Information). The force field used was AMBER*, with aqueous solvent modeled using GB/SA continuum solvation.29 The final model of the complex was obtained after unconstrained minimization to convergence. After removing DANA from the active site, compound 6 was docked using Autodock 4.2 with a 603 Å grid centered on the active site (0.375 Å resolution.)30,31 The 200 lowest energy ligand poses were evaluated by cluster analysis, and the lowest energy conformer which maintained key contacts to the active site was selected. The resulting complex was again subjected to molecular dynamics following the same protocol described above to obtain a final model of the interaction of compound 6 with NEU4. Final structures were visualized in PyMOL,32 and protein surfaces were calculated using DelPhi.33
Glossary
Abbreviations
- DANA
2-deoxy-2,3-didehydro-N-acetylneuraminic acid
- GST
glutathione S-transferase acid
- MBP
maltose binding protein
- 4MU-NANA
2′-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid
- NEU
neuraminidase
- NMR
nuclear magnetic resonance
- HRMS
high resolution mass spectrometry
- HPLC
high performance liquid chromatography
Supporting Information Available
IC50 curves, Ki determinations, and characterization of inhibitors are available in supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors acknowledge financial support from the Alberta Glycomics Centre and the Natural Sciences and Engineering Research Council of Canada. Infrastructure support was provided by the Canadian Foundation for Innovation.
The authors declare no competing financial interest.
Supplementary Material
References
- von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat. Rev. Drug Discovery 2007, 6, 967–974. [DOI] [PubMed] [Google Scholar]
- Pshezhetsky A. V.; Richard C.; Michaud L.; Igdoura S.; Wang S. P.; Elsliger M. A.; Qu J. Y.; Leclerc D.; Gravel R.; Dallaire L.; Potier M. Cloning, expression and chromosomal mapping of human lysosomal sialidase and characterization of mutations in sialidosis. Nat. Genet. 1997, 15, 316–320. [DOI] [PubMed] [Google Scholar]
- Monti E.; Bonten E.; D’Azzo A.; Bresciani R.; Venerando B.; Borsani G.; Schauer R.; Tettamanti G.. Sialidases in Vertebrates: A Family Of Enzymes Tailored For Several Cell Functions. Advances in Carbohydrate Chemistry and Biochemistry; Academic Press: 2010; Vol. 64, pp 403–479. [DOI] [PubMed] [Google Scholar]
- Pshezhetsky A. V.; Hinek A. Where catabolism meets signalling: neuraminidase 1 as a modulator of cell receptors. Glycoconjugate J. 2011, 28, 441–452. [DOI] [PubMed] [Google Scholar]
- Miyagi T.; Yamaguchi K. Mammalian sialidases: Physiological and pathological roles in cellular functions. Glycobiology 2012, 22, 880–896. [DOI] [PubMed] [Google Scholar]
- Gadhoum S. Z.; Sackstein R. CD15 expression in human myeloid cell differentiation is regulated by sialidase activity. Nat. Chem. Biol. 2008, 4, 751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyrantepe V.; Iannello A.; Liang F.; Kanshin E.; Jayanth P.; Samarani S.; Szewczuk M. R.; Ahmad A.; Pshezhetsky A. V. Regulation of Phagocytosis in Macrophages by Neuraminidase 1. J. Biol. Chem. 2010, 285, 206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen A. J. G.; Josefsson E. C.; Rumjantseva V.; Liu Q. P.; Falet H.; Bergmeier W.; Cifuni S. M.; Sackstein R.; von Andrian U. H.; Wagner D. D.; Hartwig J. H.; Hoffmeister K. M. Desialylation accelerates platelet clearance following refrigeration and initiates GPIbα metalloproteinase-mediated cleavage in mice. Blood 2011, 119, 1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dridi L.; Seyrantepe V.; Fougerat A.; Pan X.; Bonneil É.; Thibault P.; Moreau A.; Mitchell G. A.; Heveker N.; Cairo C. W.; Issad T.; Hinek A.; Pshezhetsky A. V.. Positive Regulation of Insulin Signaling by Neuraminidase 1. Diabetes 2013, in press [DOI] [PMC free article] [PubMed]
- Yamaguchi K.; Shiozaki K.; Moriya S.; Koseki K.; Wada T.; Tateno H.; Sato I.; Asano M.; Iwakura Y.; Miyagi T. Reduced Susceptibility to Colitis-Associated Colon Carcinogenesis in Mice Lacking Plasma Membrane-Associated Sialidase. PLoS One 2012, 7, e41132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster T. M.; Vocadlo D. J. Developing inhibitors of glycan processing enzymes as tools for enabling glycobiology. Nat. Chem. Biol. 2012, 8, 683–694. [DOI] [PubMed] [Google Scholar]
- Magesh S.; Moriya S.; Suzuki T.; Miyagi T.; Ishida H.; Kiso M. Design, synthesis, and biological evaluation of human sialidase inhibitors. Part 1: Selective inhibitors of lysosomal sialidase (NEU1). Bioorg. Med. Chem. Lett. 2008, 18, 532–537. [DOI] [PubMed] [Google Scholar]
- Khedri Z.; Li Y.; Cao H.; Qu J.; Yu H.; Muthana M. M.; Chen X. Synthesis of selective inhibitors against V. cholerae sialidase and human cytosolic sialidase NEU2. Org. Biomol. Chem. 2012, 10, 6112–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Cao H.; Yu H.; Chen Y.; Lau K.; Qu J.; Thon V.; Sugiarto G.; Chen X. Identifying selective inhibitors against the human cytosolic sialidase NEU2 by substrate specificity studies. Mol. BioSyst. 2011, 7, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata K.; Koseki K.; Yamaguchi K.; Moriya S.; Suzuki Y.; Yingsakmongkon S.; Hirai G.; Sodeoka M.; Von Itzstein M.; Miyagi T. Limited inhibitory effects of oseltamivir and zanamivir on human sialidases. Antimicrob. Agents Chemother. 2008, 52, 3484–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albohy A.; Mohan S.; Zheng R. B.; Pinto B. M.; Cairo C. W. Inhibitor selectivity of a new class of oseltamivir analogs against viral neuraminidase over human neuraminidase enzymes. Bioorg. Med. Chem. 2011, 19, 2817–22. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Albohy A.; Smutova V.; Pshezhetsky A.; Cairo C. W. Identification of selective inhibitors of human sialidase isoenzymes using C4, C7-modified 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA) analogs. J. Med. Chem. 2013, 56, 2948–2958. [DOI] [PubMed] [Google Scholar]
- Zou Y.; Albohy A.; Sandbhor M.; Cairo C. W. Inhibition of human neuraminidase 3 (NEU3) by C9-triazole derivatives of 2,3-didehydro-N-acetyl-neuraminic acid. Bioorg. Med. Chem. Lett. 2010, 20, 7529–7533. [DOI] [PubMed] [Google Scholar]
- Chavas L. M. G.; Tringali C.; Fusi P.; Venerando B.; Tettamanti G.; Kato R.; Monti E.; Wakatsuki S. Crystal structure of the human cytosolic sialidase Neu2—Evidence for the dynamic nature of substrate recognition. J. Biol. Chem. 2005, 280, 469–475. [DOI] [PubMed] [Google Scholar]
- Markely L. R. A.; Ong B. T.; Hoi K. M.; Teo G.; Lu M. Y.; Wang D. I. C. A high-throughput method for quantification of glycoprotein sialylation. Anal. Biochem. 2010, 407, 128–133. [DOI] [PubMed] [Google Scholar]
- Seyrantepe V.; Canuel M.; Carpentier S.; Landry K.; Durand S.; Liang F.; Zeng J.; Caqueret A.; Gravel R. A.; Marchesini S.; Zwingmann C.; Michaud J.; Morales C. R.; Levade T.; Pshezhetsky A. V. Mice deficient in Neu4 sialidase exhibit abnormal ganglioside catabolism and lysosomal storage. Hum. Mol. Genet. 2008, 17, 1556–1568. [DOI] [PubMed] [Google Scholar]
- Seyrantepe V.; Lema P.; Caqueret A.; Dridi L.; Hadj S. B.; Carpentier S.; Boucher F.; Levade T.; Carmant L.; Gravel R. A.; Hamel E.; Vachon P.; Di Cristo G.; Michaud J. L.; Morales C. R.; Pshezhetsky A. V. Mice Doubly-Deficient in Lysosomal Hexosaminidase A and Neuraminidase 4 Show Epileptic Crises and Rapid Neuronal Loss. PLoS Genet. 2010, 6, e1001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K.; Mitoma J.; Hosono M.; Shiozaki K.; Sato C.; Yamaguchi K.; Kitajima K.; Higashi H.; Nitta K.; Shima H.; Miyagi T. Sialidase NEU4 Hydrolyzes Polysialic Acids of Neural Cell Adhesion Molecules and Negatively Regulates Neurite Formation by Hippocampal Neurons. J. Biol. Chem. 2012, 287, 14816–14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki K.; Yamaguchi K.; Takahashi K.; Moriya S.; Miyagi T. Regulation of Sialyl Lewis Antigen Expression in Colon Cancer Cells by Sialidase NEU4. J. Biol. Chem. 2011, 286, 21052–21061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albohy A.; Li M. D.; Zheng R. B.; Zou C.; Cairo C. W. Insight into substrate recognition and catalysis by the mammalian neuraminidase 3 (NEU3) through molecular modeling and site directed mutagenesis. Glycobiology 2010, 20, 1127–1138. [DOI] [PubMed] [Google Scholar]
- Pshezhetsky A. V.; Potier M. Association of N-Acetylgalactosamine-6-sulfate Sulfatase with the Multienzyme Lysosomal Complex of β-Galactosidase, Cathepsin A, and Neuraminidase. J. Biol. Chem. 1996, 271, 28359–28365. [DOI] [PubMed] [Google Scholar]
- Thompson J. D.; Higgins D. G.; Gibson T. J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [DOI] [PubMed] [Google Scholar]
- Still W. C.; Tempczyk A.; Hawley R. C.; Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar]
- Goodsell D. S.; Morris G. M.; Olson A. J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.5.0.4; Schrodinger, LLC: http://www.pymol.org/ (November 15, 2012).
- Gilson M. K.; Sharp K. A.; Honig B. H. Calculating the electrostatic potential of molecules in solution: Method and error assessment. J. Comput. Chem. 1988, 9, 327–335. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.