

Abstract
We have developed a methodology that enables for the rapid measurement of ionization constants (pKa) of series of compounds by UV spectrophotometry. This protocol, which is straightforward to set up, takes advantage of the sensitivity of UV spectroscopy and the throughput enabled by the 96-well microplate (as opposed to the use of 1 cm quartz cuvette). The compounds, in stock solutions in DMSO, are dissolved in several aqueous buffer solutions directly in the microtiter plate, allowing the simultaneous determination of the UV spectra as a function of pH. Further treatment of the data provides the pKa values in a medium-throughput manner. The pKa values of 11 new antitrypanosomal dibasic compounds were determined using this methodology.
Keywords: pKa, ionization constant, imidazoline, 96-well microplate, UV spectrophotometry, buffer solution, trypanosome, sleeping sickness, dibasic compounds
The ionization constant (pKa) of a drug is a physicochemical parameter that significantly affects its pharmacokinetic behavior, that is, absorption, distribution, metabolism, and excretion (ADME). Lipophilicity, solubility, protein binding, and permeability of a compound are influenced by its pKa;1 for instance, basic compounds with pKa ≥ 7.4 will be charged at physiological pH and display slower diffusion rate across biological membranes such as the blood–brain barrier (BBB).
In the last years, we have been interested in the design of new antitrypanosomal dicationic compounds as potential agents against late-stage sleeping sickness.2−4 This disease caused by subspecies of African trypanosomes is characterized by an early hemolymphatic stage that is followed by a fatal late stage due to an invasion of the CNS by the parasites. In the course of this project, we have designed new derivatives of dibasic lead compounds modulating the basicity of the nitrogen moieties with O-alkyl substituents to improve their BBB permeability. Thus, measuring the pKa of these new series was a necessary task to rationalize the in vitro and in vivo biological activity findings.
Different methods exist to measure pKa values,5 the following ones being the most common: (a) potentiometric titration, where the pKa is derived from the titration curve; (b) spectrophotometric titration, where a UV spectrum of the compound is taken for each point of the titration and the change in UV absorbance is plotted against the pH;6 and (c) capillary electrophoresis in different buffer solutions with results calculated from the relative mobility of ions at different pH.7−11 Among these, the potentiometric method requires a larger quantity of sample as compared to UV spectroscopy or capillary electrophoresis. In addition, special equipment has been developed for measuring pKa in an automatic way using a mixed-buffer linear pH gradient system.12,13 However, in the absence of such an apparatus, the determination of pKa for series of compounds by traditional methods can be a time-consuming task.
In the present paper, we report an efficient medium-throughput method for the determination of ionization constants by UV spectroscopy using standard 96-well microtiter plates. This methodology, which is straightforward to set up, takes advantage of the sensitivity of UV spectroscopy (i.e., only a small amount of compound is needed, useful for sparingly soluble substances, work at high and low pH values)5 and the throughput enabled by the 96-well microplate. It does not require complex equipment (apart from a UV spectrophotometer equipped with a microplate reader and a pH meter for the preparation of the buffer solutions) and allows for the rapid measurement of pKa values of series of compounds.
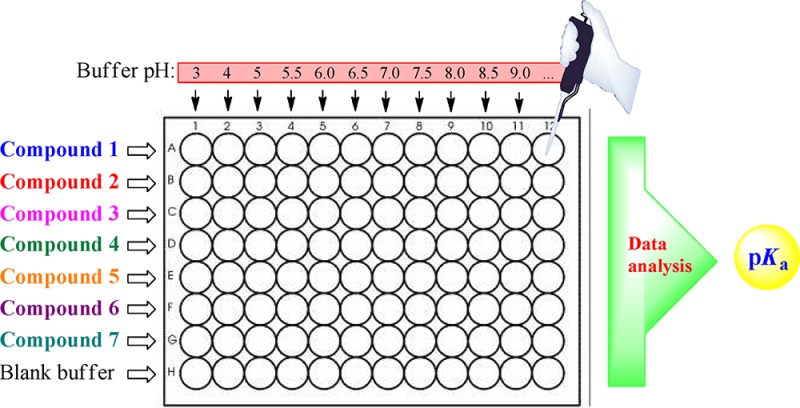
To measure pKa values by UV spectrophotometry, compounds must have a chromophore close to the ionization centers, and the spectrum (absorbance) must change as a function of ionization. Briefly, our method proceeds as follows: (1) the compounds under study are prepared as 10 mM stock solutions in DMSO; (2) the 96-well microtiter plate is filled with different buffers of constant ionic strength (I = 0.1 M) ranging from pH 3 to pH 12 (e.g., 2 points per pH unit); (3) a fixed amount of compound stock solution is added to each well, leaving a series of blank buffers (i.e., free of compound) as correction factors (note that the final amount of DMSO in the well is ≤2% v/v); (4) the UV spectra of the compounds in the range 230–500 nm are recorded; and (5) the data of the UV spectra at each wavelength as a function of pH and the pKa are processed using the Prism program.14
In the present study, we have used a similar data analysis as described by Tomsho et al. for the determination of ionization constants by spectral analysis (see the Supporting Information for details).15 Figure 1 shows an example of such an analysis for the determination of the pKa of 4-nitrophenol. The changes observed in the UV spectrum of 4-nitrophenol depending on the species present in solution are clearly seen in Figure 1. At pH 3, the molecule is neutral (4-NO2PhOH) with a maximum absorption at 314 nm, whereas at pH 10, only the deprotonated species is present (4-NO2PhO–) with λmax ≈ 400 nm. The pKa measured in H2O at 30 °C (7.02 ± 0.01) was in good agreement with the reported value (7.16 at 25 °C) (Table 1).
Figure 1.
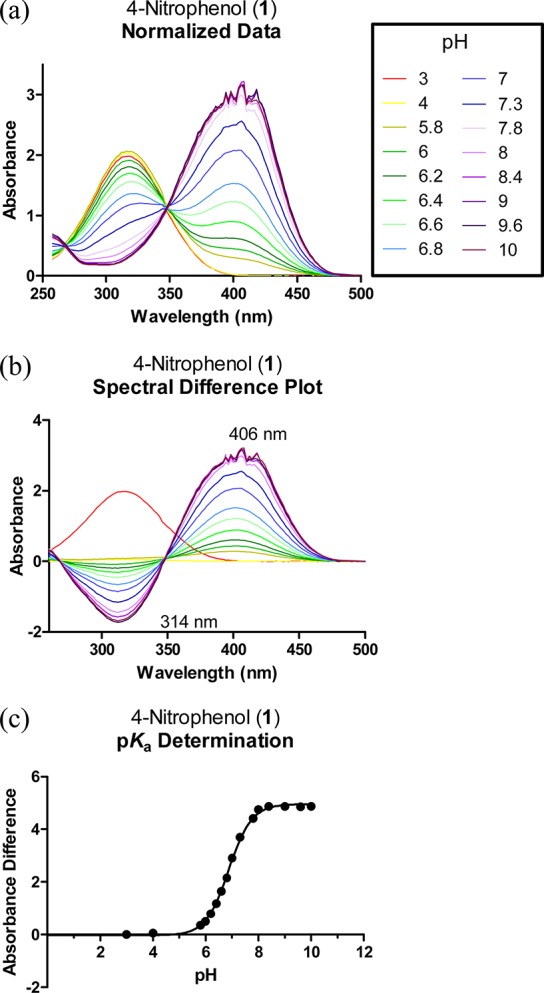
Spectral data analysis and pKa determination. (a) UV spectrum (λ = 250–500 nm) of 4-nitrophenol (1) in different aqueous buffer solutions ranging from pH 3 to 10. The absorbances are normalized to zero for λ = 500 nm. (b) Plot of the spectral difference between different solutions of 1. The maximum positive deviation occurred at 406 nm, while the maximum negative deviation occurred at 314 nm. (c) Plot of the total absorbance difference vs pH to determine the pKa. The total absorbance difference is the sum of the absolute absorbance difference values at the chosen wavelengths (i.e., 314 and 406 nm). The pKa value was worked out by nonlinear regression using eq 1 according to Tomsho et al. (see the Experimental Procedures).15
Table 1. pKa Values of Monoacidic, Monobasic, and Dibasic Compounds Determined by the 96-Well UV Spectrophotometric Method.

The use of 2% v/v DMSO as a cosolvent did not alter significantly the pKa value of the test compounds. Working temperature = 30 °C. All pKa values were measured at constant ionic strength (I = 0.1 M) and concentration (C = 0.2 mM).
Analytical wavelengths are determined at the maximum and minimum absorption values in the spectral difference plot.
Experiments were repeated at least three times.
Standard deviation.
Experimental pKa values at 25 °C in water.
To validate this method, the pKa values of five compounds previously reported in the literature were measured using our protocol with an average of 16 different buffer solutions ranging ±2 pH units around the pKa value (i.e., 16 points in the absorbance vs pH curve). As shown in Table 1, the observed pKa values were in good agreement with the reported data. We could not discriminate two different pKa values for the symmetrical dicarbamimidate derivative 5. This is consistent with earlier report by Nagle et al.,16 who measured only one ionization constant (pKa = 10.4) for both basic groups by acid–base titration using UV spectroscopy.
We have checked that the use of 2% v/v DMSO as a cosolvent did not alter significantly the pKa value of the tested compounds. This is an advantage as poorly water-soluble compounds can be tested using this protocol. Finally, we have observed that repeating the experiment three times for each compound gave, in most cases, satisfactory scatter values. (Scatter, as defined by Albert and Serjeant,5 is the largest deviation between any value in the set and the value calculated by taking antilogarithms of each pKa value in a set, averaging these, and writing down the logarithm of the average as the pKa.)
Experimentally, the time needed to load manually the 96-well microplate (e.g., blank buffer + seven different compounds in 12 buffers or five compounds in 16 buffers) is about 45 min, whereas the reading of the plate takes approximately 40 min (e.g., with a 2 nm resolution). We have checked that the compounds solutions were stable over time and that UV spectra were not significantly affected over the course of the experiment. To do so, several readings of the same plate were repeated over a 3 h period, and the pKa was calculated at three different time points (Figure 2).
Figure 2.
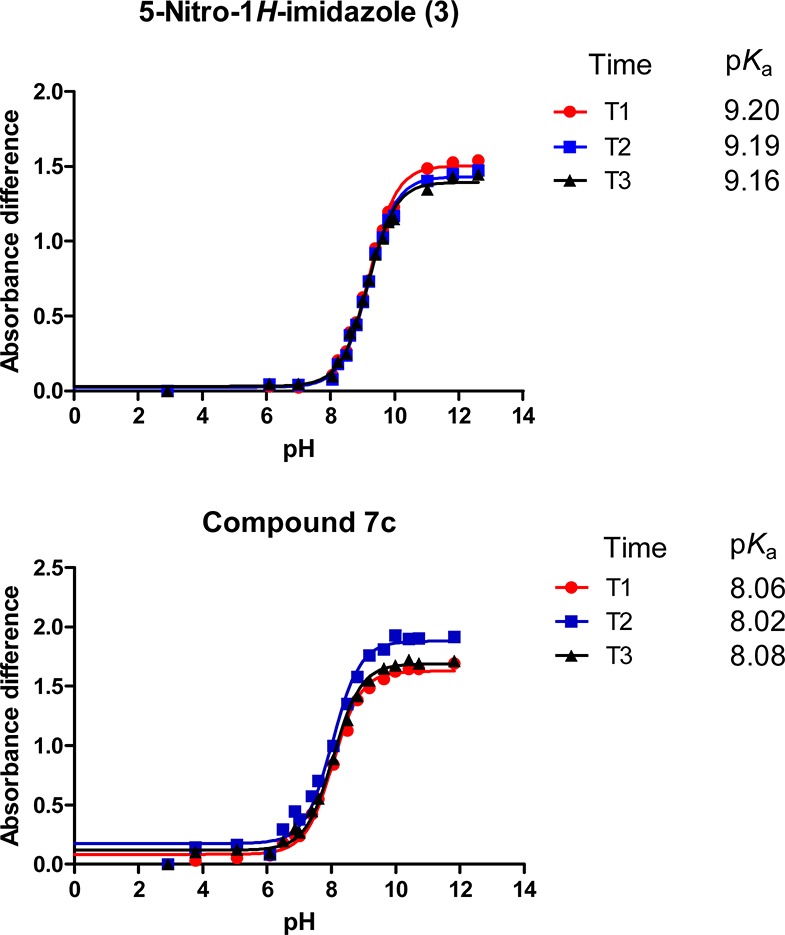
Study of the stability of the compounds in buffer solutions over time. The plates with the compounds dissolved in buffer solutions were read three times over a 3 h period (T1 = 40 min, T2 = 90 min, and T3 = 150 min). The pKa was calculated for each time point.
The pKa values of a series of 11 dibasic compounds synthesized in our group as antitrypanosomal agents were measured in triplicate using this methodology (Table 2). It should be noted that only one pKa value could be determined, suggesting overlapping pKa values for both imidazoline groups. This is possibly due to the second deprotonation taking place in a very small pH range so that changes in the spectra cannot be distinguished. Rozas and co-workers also failed to distinguish two discrete pKa of similar dibasic compounds.16,17 It should be noted that in our particular case, that is, symmetrical dibasic compounds, the scatter values were higher (0.03–0.2) than in compounds having only one basic site (0.01–0.1). This may be attributed to the fact that the bisimidazolines have overlapping pKa values or to possible solubility problems giving rise to product precipitation in the wells. These results show that the introduction of a hydroxyl group at N1-nitrogen of the imidazoline ring reduces its ionization constant by approximately 2 pKa units (compare 6b/6a and 7b/7a). Introduction of other alkoxy substituents (OMe and OEt) at the same position does not alter significantly the pKa as compared to a hydroxyl group.
Table 2. Ionization Constants of New Dibasic Antitrypanosomal Compounds Determined by the 96-Well UV Spectrophotometric Method at 30 °C and Constant Ionic Strength (I = 0.1 M).
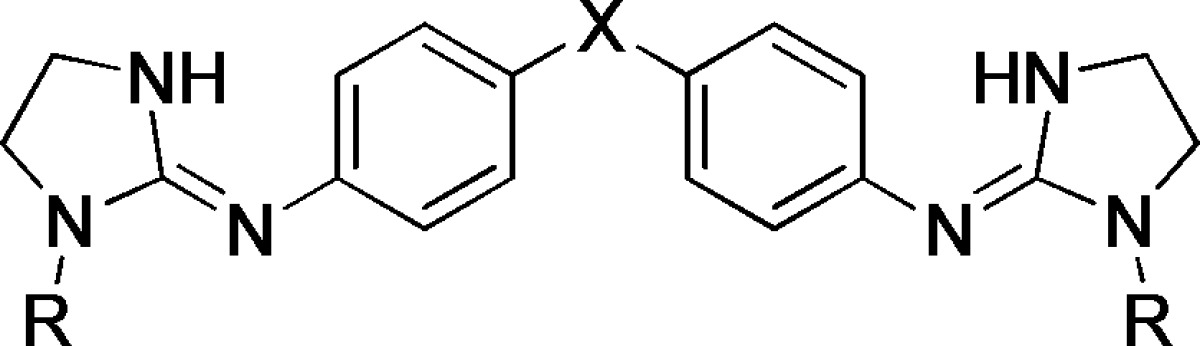
| compda | X | R | λ (nm)b | pKac |
|---|---|---|---|---|
| 6a | NHCO | H | 258/308 | 9.29 ± 0.07d |
| 6b | OH | 258/298 | 7.43 ± 0.10 | |
| 6c | OMe | 258/312 | 7.27 ± 0.09 | |
| 6d | OEt | 258/308 | 7.34 ± 0.03 | |
| 7a | CH2CH2 | H | 260/310 | 10.71 ± 0.10 |
| 7b | OH | 238/264 | 7.97 ± 0.13 | |
| 7c | OMe | 236/262 | 8.01 ± 0.12 | |
| 7d | OEt | 238/262 | 8.01 ± 0.21 | |
| 8a | NHCONH | H | 260/292 | 10.34 ± 0.04 |
| 8c | OMe | 240/270 | 7.95 ± 0.05 | |
| 8d | OEt | 258/296 | 8.27 ± 0.07 |
The compounds were dissolved in DMSO (stock solution) and diluted with the corresponding buffers to reach a final concentration of 0.2 mM in the well (the final quantity of DMSO is 2% v/v).
Analytical wavelength.
pKa of the aminoimidazoline groups; only one pKa could be calculated for both imidazolines.
The pKa values are the mean of three or four (for 7c and 8d) independent measurements ± SDs.
In summary, we present here a very convenient method to measure ionization constants by UV spectrophotometry in a medium-throughput manner. Satisfactory pKa values can be obtained with a minimum of three repeats for each experiment. We trust that this methodology will be of interest to medicinal chemists when assessing pKa values of medium-size compound libraries for the study of their structure–activity relationships.
Experimental Procedures
The buffer solutions used here were a follows: (a) AcOH/AcONa, covering the pH range 3.0–5.0; (b) KH2PO4/K2HPO4, covering the pH range 6.0–8.0; (c) Borax/HCl, covering the pH range 8.2–9.0; (d) Borax/NaOH, covering the pH range 9.2–10.8; (d) Na2HPO4/Na3PO4, covering the pH range 10.8–12; and (e) glycine/NaOH for pH 12.6. All of these buffer solutions were prepared with the same ionic strength (I = 0.1 M) by adding KCl (see the Supporting Information for details). The stock solutions of the compounds were prepared at 10 mM concentration in DMSO. Standard solutions were prepared adding 4 μL of the stock solution in 196 μL of buffer solution in each well of the microplate getting a final concentration of 0.2 mM. In some cases where saturated spectra were observed, 0.1 mM analyte solutions were used (e.g., 2 μL of stock solution in 198 μL of buffer). UV spectra were recorded on a THERMO Multiskan Spectrum apparatus between 210 and 400 nm (230–500 nm for compound 1) at 2 nm resolution. The data were processed with the Excel program. The pKa values were worked out by nonlinear regression (Prism program) using eq 1 as reported.15
| 1 |
εHA and εA– are the extinction coefficients of the acid and base forms of the compound, respectively (i.e., the minima and maxima of the absorbance difference curve, respectively), and [St] is the total compound concentration. See the Supporting Information for a detailed description of the experimental protocols and data analysis.
Acknowledgments
We thank Dr. Rozas for useful comments on this manuscript and for providing us with the reference compound 5.
Glossary
Abbreviations
- BBB
blood–brain barrier
- ADME
absorption, distribution, metabolism, and excretion
Supporting Information Available
Detailed description of the experimental procedures: preparation of buffer solutions, loading of the 96-well microtiter plate, data analysis, multiwavelengths spectra, and pKa determination plots for compounds 2–8. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. C.D. and C.H.R.M. designed the experiments and analyzed the data. C.H.R.M. performed all of the experimental work.
Funding was from the Spanish Ministerio de Ciencia e Innovación (Grant SAF2009-10399). C.H.R.M. was recipient of a Ph.D. fellowship for the government of Panama (SENACYT Grant BIDP-2008-030).
The authors declare no competing financial interest.
Supplementary Material
References
- Manallack D. T. The pKa Distribution of Drugs: Application to Drug Discovery. Perspect. Med. Chem. 2007, 1, 25–38. [PMC free article] [PubMed] [Google Scholar]
- Nieto L.; Mascaraque A.; Miller F.; Glacial F.; Ríos Martínez C.; Kaiser M.; Brun R.; Dardonville C. Synthesis and Antiprotozoal Activity of N-Alkoxy Analogues of the Trypanocidal Lead Compound 4,4′-Bis(imidazolinylamino)diphenylamine with Improved Human Blood-Brain Barrier Permeability. J. Med. Chem. 2011, 54, 485–494. [DOI] [PubMed] [Google Scholar]
- Rodríguez F.; Rozas I.; Kaiser M.; Brun R.; Nguyen B.; Wilson W. D.; García R. N.; Dardonville C. New Bis(2-aminoimidazoline) and Bisguanidine DNA Minor Groove Binders with Potent in Vivo Antitrypanosomal and Antiplasmodial Activity. J. Med. Chem. 2008, 51, 909–923. [DOI] [PubMed] [Google Scholar]
- Dardonville C.; Barrett M. P.; Brun R.; Kaiser M.; Tanious F.; Wilson W. D. DNA binding affinity of bisguanidine and bis(2-aminoimidazoline) derivatives with in vivo antitrypanosomal activity. J. Med. Chem. 2006, 49, 3748–3752. [DOI] [PubMed] [Google Scholar]
- Albert A.; Serjeant E. P.. The Determination of Ionization Constants: A Laboratory Manual; Chapman and Hall: London, 1971. [Google Scholar]
- Allen R. I.; Box K. J.; Comer J. E. A.; Peake C.; Tam K. Y. Multiwavelength spectrophotometric determination of acid dissociation constants of ionizable drugs. J. Pharm. Biomed. Anal. 1998, 17, 699–712. [DOI] [PubMed] [Google Scholar]
- Fuguet E.; Rafols C.; Roses M. A fast high throughput method for the determination of acidity constants by capillary electrophoresis. 3. Basic internal standards. J. Chromatogr., A 2011, 1218, 3928–3934. [DOI] [PubMed] [Google Scholar]
- Cabot J. M.; Fuguet E.; Ràfols C.; Rosés M. Fast high-throughput method for the determination of acidity constants by capillary electrophoresis. II. Acidic internal standards. J. Chromatogr., A 2010, 1217, 8340–8345. [DOI] [PubMed] [Google Scholar]
- Fuguet E.; Ràfols C.; Bosch E.; Rosés M. Fast high-throughput method for the determination of acidity constants by capillary electrophoresis: I. Monoprotic weak acids and bases. J. Chromatogr., A 2009, 1216, 3646–3651. [DOI] [PubMed] [Google Scholar]
- Gong X.; Figus M.; Plewa J.; Levorse D. A.; Zhou L.; Welch C. J. Evaluation of multiplexed CE with UV detection for rapid pKa estimation of active pharmaceutical ingredients. Chromatographia 2008, 68, 219–225. [Google Scholar]
- Zhou C.; Jin Y.; Kenseth J. R.; Stella M.; Wehmeyer K. R.; Heineman W. R. Rapid pKa estimation using vacuum-assisted multiplexed capillary electrophoresis (VAMCE) with ultraviolet detection. J. Pharm. Sci. 2005, 94, 576–589. [DOI] [PubMed] [Google Scholar]
- Comer J.; Box K. High-Throughput Measurement of Drug pKa Values for ADME Screening. J. Lab. Autom. 2003, 8, 55–59. [Google Scholar]
- Box K.; Bevan C.; Comer J.; Hill A.; Allen R.; Reynolds D. High-Throughput Measurement of pKa Values in a Mixed-Buffer Linear pH Gradient System. Anal. Chem. 2003, 75, 883–892. [DOI] [PubMed] [Google Scholar]
- Lead A. M.; Tarkany O.; Novikov A.; Moskalenko E.; Vysotsky A.; Volokh V.. Prism 5 for Windows, 5.01; GraphPad Software Inc.: 1992–2007. [Google Scholar]
- Tomsho J. W.; Pal A.; Hall D. G.; Benkovic S. J. Ring Structure and Aromatic Substituent Effects on the pKa of the Benzoxaborole Pharmacophore. ACS Med. Chem. Lett. 2012, 3, 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle P.; Kahvedžić A.; McCabe T.; Rozas I. On the protonated state of amidinium-like diaromatic derivatives: X-ray and UV studies. Struct. Chem. 2011, 1–9. [Google Scholar]
- Kinsella G. K.; Rodriguez F.; Watson G. W.; Rozas I. Computational approach to the basicity of a series of [alpha]1-adrenoceptor ligands in aqueous solution. Bioorg. Med. Chem. 2007, 15, 2850–2855. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.