

Abstract

Takeda G-protein-coupled receptor 5 (TGR5) represents an exciting biological target for the potential treatment of diabetes and metabolic syndrome. A new class of high-throughput screening (HTS)-derived tetrahydropyrido[4,3-d]pyrimidine amide TGR5 agonists is disclosed. We describe our effort to identify an orally available agonist suitable for assessment of systemic TGR5 agonism. This effort resulted in identification of 16, which had acceptable potency and pharmacokinetic properties to allow for in vivo assessment in dog. A key aspect of this work was the calibration of human and dog in vitro assay systems that could be linked with data from a human ex vivo peripheral blood monocyte assay that expresses receptor at endogenous levels. Potency from the human in vitro assay was also found to correlate with data from an ex vivo human whole blood assay. This calibration exercise provided confidence that 16 could be used to drive plasma exposures sufficient to test the effects of systemic activation of TGR5.
Keywords: GPCR, TGR5, agonist, diabetes, GLP-1
TGR5 (Takeda G-protein-coupled receptor 5) is a class A G-protein-coupled receptor (GPCR) expressed in liver, skeletal muscle, intestine, brown adipose tissues, and monocytes.1−4 Activation of the TGR5 receptor upon ligand binding results in Gαs-coupled activation of adenylate cyclase. The subsequent downstream signaling cascade is believed to drive multiple effects that are cell type-dependent including (1) enhanced glucagon-like peptide-1 (GLP-1) release from intestinal cells potentially offering improved glycemic control through potentiation of glucose-dependent insulin secretion;5 (2) nuclear effects, such as endothelial nitric oxide synthase activation and increased nitric oxide production in liver cells;6 and (3) reduction of macrophage inflammation and lipid loading leading to inhibition of atherosclerosis.7,8 Activation of TGR5 can also induce type 2 iodothyronine deiodinase in brown adipose tissue. In mice, this leads to increased energy expenditure, but the picture is less clear for humans.9,10 For these reasons, TGR5 agonists may be useful agents to not only treat diabetes with concurrent management of glucose levels and body weight but also potentially address other aspects of metabolic syndrome.
Bile acids are the purported endogenous agonists for TGR5. Of the several bile acids that act as ligands, taurine-conjugated lithocholic acid, lithocholic acid, deoxycholic acid, chenodeoxycholic acid, and cholic acid have all been shown to dose dependently induce accumulation of cyclic adenosine 5′-monophosphate (cAMP) in TGR5-transfected Chinese hamster ovary (CHO) cells.11 Significant effort has been devoted to synthetic bile acids, such as INT-777 (1).12,13 In addition, several other nonbile acid agonists have been reported in the literature (2–4),14−16 which offer the potential for higher selectivity over other bile acid-mediated pathways (Figure 1). Previous work with 1 demonstrated in vivo modulation of GLP-1 secretion and increased energy expenditure in a diet-induced obese mouse model. Furthermore, 2 was shown to potentiate GLP-1 secretion in an in vivo dog model when dosed in an intracolonic manner and coadministered with a glucose challenge. However, in view of the physicochemical property space and data generated from evaluation of these ligands, few appeared to offer the chance of attaining reasonable free drug concentrations in animal models to allow for the assessment of the full range of systemic effects associated with activation of TGR5. Thus, our goal for this work was the identification of a suitable compound capable of fully testing the effects of systemic TGR5 agonism to progress to in vivo studies. Because the potency of GPCR agonists in overexpressed cell systems is heavily system-dependent, we were very mindful to set appropriate goals for the level of TGR5 potency required in our tool compound.17 Thus, an additional goal for this work became the establishment of a quantitative pharmacological relationship between biological systems that express TGR5 at endogenous levels and overexpressed assay systems commonly used in the literature.18 Furthermore, we set the objective that the “endogenous” assay system used for this calibration should be one with an output linked to a clinically relevant end point [e.g., tumor necrosis factor-α (TNF-α)].
Figure 1.

Previously disclosed TGR5 agonists.
Literature reports and our evaluation of receptor homology among potential preclinical species suggested that careful selection of the animal model for in vivo evaluation would be required. Specifically, the relatively low homology between human and rodent TGR5 receptors coupled with the divergent structure–activity relationships (SARs) of human and rodent TGR5 activity for nonbile acid agonists15 suggested that a nonrodent preclinical species would be necessary. Dog TGR5 agonist activity, however, is generally in line with human TGR5 agonist activity.14 Thus, we sought to identify a potent, orally available TGR5 dog agonist that, if successful, might also act as a lead for a human agent. A key requirement would be the ability to rigorously test the safety and efficacy of systemic TGR5 activation by a nonbile acid agonist. Our medicinal chemistry effort toward such a tool compound is described herein.
A suitable starting point for generation of a tool compound was identified through high-throughput screening (HTS) of the Pfizer compound collection. The HTS assay system was tuned for high sensitivity and a large dynamic range, which is ideal for screening compounds with a wide variation in potency. In this case, a doxycycline-regulated promoter system was used to overexpress the human TGR5 receptor in a CHO cell line (induced assay). Elevation of cAMP was used as the detection system. A previously undisclosed chemotype, as represented by compound 5, was discovered using this process. The MW, Log D, and topological polar surface area (TPSA) were acceptable, as were permeability (Ralph Russ canine kidney, RRCK)19 and safety {[3H]-dofetilide binding, cytochrome P450 (CYP) inhibition} assessments. However, 5 showed very high intrinsic clearance in human and rat liver microsomes (HLM and RLM), indicating that significant effort would be required to achieve a reasonable clearance profile that could translate into a systemically available tool compound. Early on, a small cohort of compounds, including 5, was subjected to further in vitro characterization in mouse-, rat-, and dog-induced cell lines, thus confirming the similarity between dog/human activity and the disparity for rodent/human activity for this series (Table 1).
Table 1. Pharmacological, Absorption, Metabolism, and Safety Properties of 5.
| 5 (R1 = EtNH, R2 = iPr) | |
|---|---|
| MW 395.5, eLog D 3.40, TPSA 101 | |
| hTGR5 induced EC50 (nM) | 2.5 |
| mTGR5 induced EC50 (nM) | 5620 |
| rTGR5 induced EC50 (nM) | 3930 |
| dTGR5 induced EC50 (nM) | 6 |
| RRCK (Papp, 10–6 cm/s)a | 20.2 |
| RLM CLint (μL/min/mg) | >564 |
| HLM CLint (μL/min/mg) | >317 |
| CYP (pct inh at 3 μM) | 1A2, 2D6, 2C9, 3A4 < 20% |
| [3H]-dofetilide (pct inh at 10 μM) | 9% |
RRCK = modified Madin–Darby permeability assay.
With lead 5 in hand, a screening cascade was implemented to facilitate identification of an orally bioavailable tool compound suitable for in vivo dog studies. Because both TGR5 agonist activity and metabolic fate in liver microsomes for human and dog were found to be generally aligned, the screening cascade initiated with parallel assessment of activity (human TGR5, induced assay) and HLM stability as the first decision points. Compounds with higher potency and lower intrinsic clearance relative to 5 were next profiled against in vitro cAMP assays described above without doxycycline (uninduced assay)20 for both human and dog TGR5, as well as assessment of clearance in dog liver microsomes (DLM). Thus, the uninduced assay is based on cells expressing basal receptor numbers. The initial optimization process was designed to improve clearance by reduction of lipophilicity, minimize metabolic soft spots, and maximize TGR5 potency in the induced assay.
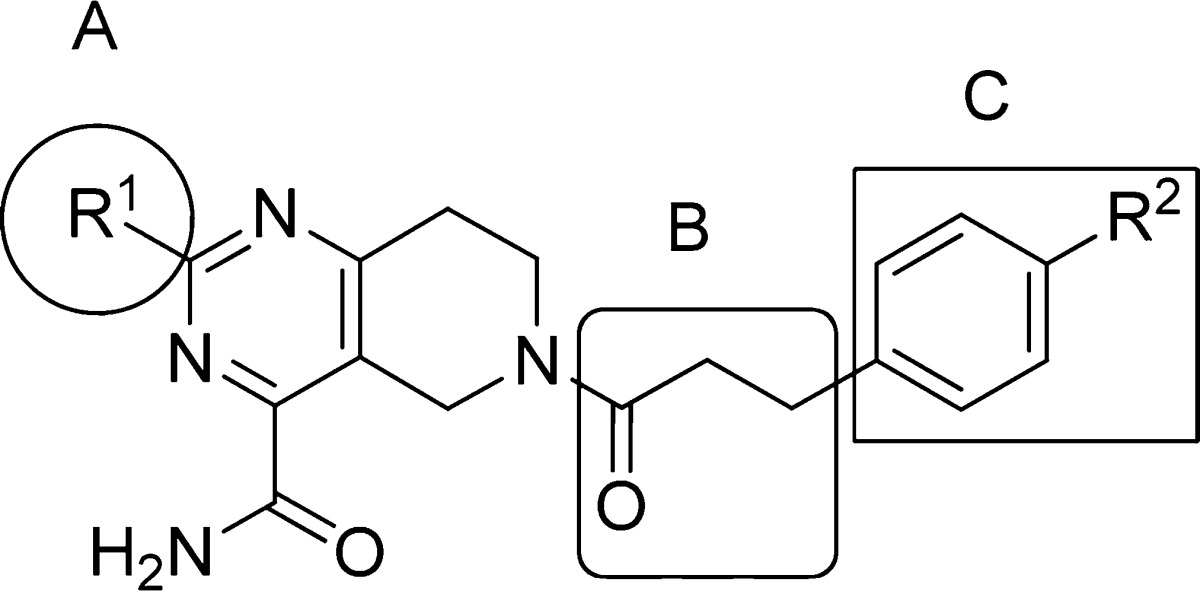
Compound 5 was divided into three fragments: the 2-substituent of the pyrimidine (A), the acyl linker (B), and the aryl group (C) for the purpose of exploring the SARs. Construction of the various tetrahydropyrido[4,3-d]pyrimidine cores is shown in Scheme 1. The enamine generated from N-Boc 4-piperidinone 6 was acylated with ethyl oxalyl chloride to give 7, then cyclized with an amidine, urea, or 2-methylisothiourea to provide the pyrimidines 8. Subsequent manipulation of the 2-substituent of the pyrimidine followed by deprotection provided 9 with a range of small alkyl, alkylamino, or ether groups at R1. Variation of the amide substituent, using library or singleton chemistry, allowed for combination of cores with different R1 groups with a diverse set of acids. These were coupled under standard amide bond formation conditions [O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) or 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI)]. Expansion of the fragment C region was performed using two complementary, two-step parallel chemistry procedures. As outlined in Scheme 2, common intermediates 10 or 11 were combined with aldehydes (Horner–Emmons–Wadsworth) or aryl halides (Heck) followed by reduction to furnish the desired analogues. The library-based strategy, described above, allowed for modulation of the linker and aryl moieties to rapidly assess the B/C region of the molecule on a defined range of 2-substituted tetrahydropyrido[4,3-d]pyrimidine cores.
Scheme 1. Synthesis of Tetrahydropyrido[4,3-d]pyrimidine Core(s).

Reagents and conditions: PG = Boc or Cbz. (a) Morpholine, toluene, 110 °C. (b) Ethyl oxalyl chloride, Et3N, CH2Cl2. (c) R1-amidine, 2-methyl-isothiourea or urea, Et3N, EtOH, reflux. (d) Ammonia, MeOH. (e) Conversion of R1 = SMe into R1 = EtNH: (1) 3-chloroperoxybenzoic acid, CH2Cl2; (2) EtNH2, THF. (f) Conversion of R1 = OH into R1 = Cl: POCl3, acetone. (g) Conversion of R1 = Cl into R1 = cPr: cPrZnBr, PEPPSITM-IPr,21 THF. (h) Conversion of R1 = Cl into R1 = OEt: NaH/EtOH. (i) 4 N HCl, dioxane. (j) Pd/C, H2, MeOH.
Scheme 2. Synthesis of Phenpropanamides.

Reagents and conditions: (a) Aldehyde, base. (b) Et3SiH, EtOH, cat. 5% Pd/C. (c) Aryl halide, (tBu3P)2Pd, dioxane, 95 °C.
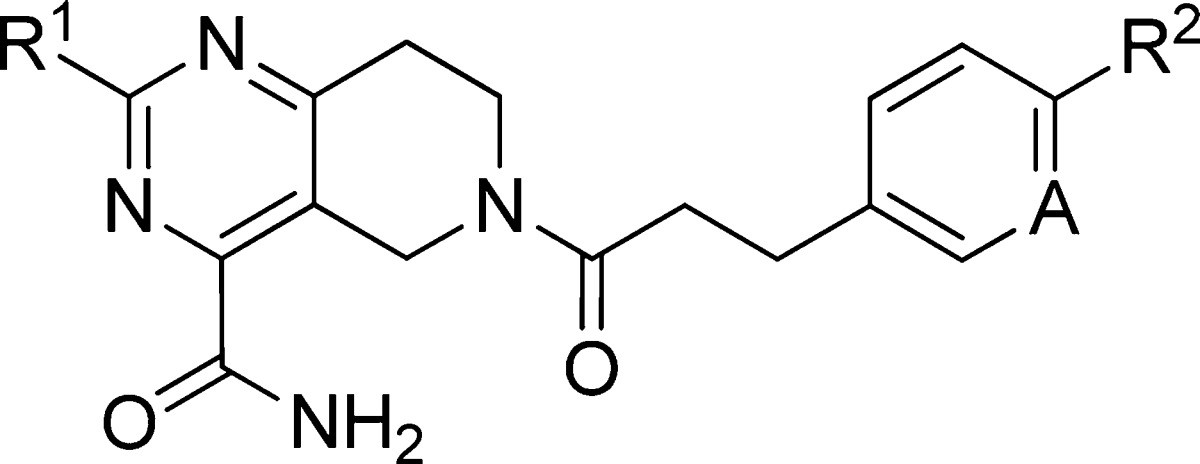
The SAR trends for changes to the core and the B/C region could be gleaned from these libraries and are represented by selected compounds shown in Table 2. Compounds that were potent (EC50 < 10 nM) in the induced assay were advanced to screening in the uninduced version of the human and dog assays. The uninduced EC50 values for all analogues were right-shifted 50–150-fold relative to the induced EC50 values as anticipated from standard receptor theory.17 The correlation between human and dog potency was maintained. Even in the uninduced assay system, which should allow greater differentiation of agonist efficacy, all compounds were full agonists relative to control.22 In general, minor changes to the original structure resulted in large shifts in agonist potency. We hypothesize that minor structural changes can alter the optimal agonist conformation of the ligand resulting in functionally significant changes to receptor conformation. Aromatic moieties linked by an ethylene spacer to the amide carbonyl were highly preferred. Changes made to the linking ethylene group (truncation, heteroatom interruption, and alkyl substitution) resulted in compounds with diminished potency (data not shown). Para substitution (R2) of the aromatic ring (phenyl, pyridyl) was preferred. Significant boosts in potency were noted in increasing the size of the para substituent from H (12) to CH3 (13) and then CH3CH2 (14). Resulting increases in microsomal clearance from these structural changes correlate with higher Log D values. The potency increase in adjustment of the para substituent from OMe (15) to OCF3 (16) also warrants further discussion. Conformational analysis revealed that the ethyl of 14 and the OCF3 of 16 lie orthogonal to the plane of the phenyl ring, while the methoxy of 15 lies in the plane of the phenyl,23 suggesting that the out of plane substituent could contribute to attaining a favorable agonist conformation. Additional fluoro-containing para substituents provided 17–19 that were equipotent with 16 but, despite having lower lipophilicity, suffered from high metabolic clearance. Incorporation of a pyridine led to a greater reduction of Log D for 20. However, the resulting improvement in microsomal clearance for 20 came at the expense of a 20–40-fold decrease in potency relative to 19 and 16, respectively. Conservative changes to the pyrimidine substituent (R1) to give 21 (Et), 22 (EtO), and 23 (cPr) were tolerated. More extensive changes to R1, smaller or larger, usually resulted in an order of magnitude drop in potency and provided no metabolic stability advantage (data not shown).
Table 2. Potency and Properties of Selected Analoguesa.
| EC50 (nM)b |
CLintc |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | A | induced hTGR5 | uninduced hTGR5 | uninduced dTGR5 | HLM | DLM | eLog D |
| 1 | 166 | 5540 | 4960 | <8.0 | 1.85d | ||||
| 2 | 24 | 316 | 445 | >320 | 3.90 | ||||
| 3 | 154 | 3220 | 2060 | 249 | 5.10 | ||||
| 4 | 47 | 1420 | 584 | >320 | 630 | 2.90 | |||
| 12 | EtNH | H | CH | 257 | 8320 | N.T. | 79 | 1.40 | |
| 13 | EtNH | CH3 | CH | 78 | 3780 | 5800 | 168 | 2.00e | |
| 14 | EtNH | CH2CH3 | CH | 9.2 | 215 | 828 | 238 | 2.40 | |
| 5 | EtNH | CH(CH3)2 | CH | 3.6 | 158 | 213 | >317 | 139 | 3.40 |
| 15 | EtNH | OCH3 | CH | 137 | 2430 | 4280 | 96 | 27 | 1.05 |
| 16 | EtNH | OCF3 | CH | 2.3 | 75 | 135 | 184 | 115 | 3.00 |
| 17 | EtNH | SCF3 | CH | 1.6 | 48 | 112 | >298 | 3.50 | |
| 18 | EtNH | CH2CF3 | CH | 4.7 | 154 | 233 | 141 | 68 | 2.60 |
| 19 | EtNH | CF2CH3 | CH | 2.7 | 146 | 382 | 211 | 2.20 | |
| 20 | EtNH | CF2CH3 | N | 43 | 2990 | 3200 | 18 | 18 | 1.40 |
| 21 | Et | OCF3 | CH | 2.0 | 83 | 220 | 174 | 2.90 | |
| 22 | EtO | OCF3 | CH | 3.3 | 112 | 303 | 115 | 2.90 | |
| 23 | cPr | OCF3 | CH | 2.8 | 43 | 407 | 217 | 3.30 | |
NT, not tested.
Most values are reflective of n ≥ 3 experiments. Please see the Supporting Information for the number of experiments and associated error. All values are for full agonists relative to a standard.
Intrinsic clearance μL/min/mg.
Measured Log D by shake-flask method.
Calculated Log D using a model based on Cubist modeling methodology [Rulequest Research, www.rulequest.com]. The training set for the model was based on >45000 measured eLog D values.24
With potent TGR5 agonists against both induced and uninduced assays in hand, we next sought to establish a correlation between the primary cAMP assays in cells recombinantly expressing human TGR5 with human cell lines that natively express TGR5. This would serve to improve confidence in functional potency determinations and ensure that adequate levels of TGR5 potency had been achieved within our series. Compound 16 was selected for characterization in human NCI-H716 cells (gut-derived) and peripheral blood mononuclear cells (PBMCs) isolated from whole human blood to assess its TGR5 cAMP activities (Table 3). Of note, an apparent correlation between the potency from the uninduced assay with the potencies from NCI-H716 cell and ex vivo human PBMC cAMP assays was established, while the induced assay appears to significantly overpredict potency.
Table 3. Activation of TGR5 with 16 in Multiple Assay Systems.
| 16 | EC50 (nM)a |
|---|---|
| induced hTGR5 | 2.3 ± 1.5 |
| uninduced hTGR5 | 75 ± 72 |
| NCI-H716 (cAMP) | 145 ± 73 |
| ex vivo PBMC (cAMP) | 110 ± 67 |
Values are means of >3 experiments ± standard deviations.
An ex vivo whole blood assay was also developed. After a 30 min compound pretreatment, stimulation of human whole blood with lipopolysaccharides (LPS, 10 ng/mL, 4–5 h of stimulation), the suppression of TNF-α release in a dose-dependent manner was demonstrated with compound 16 (Figure 2). Similar results were noted for a compound from a distinct and highly dissimilar chemotype.25
Figure 2.

Inhibition of TNF-α secretion, ex vivo whole blood LPS challenge. Data based on an average of three donors. Normalized to maximum TNF-α secreted.
Having established a correlation between our uninduced TGR5 assay and pharmacology in native systems, we then turned our attention toward identifying a tool compound for use in vivo that may be able to achieve levels of exposure consistent with these findings. To better understand the clearance profiles of compounds 14 and 16, in vitro metabolite identification studies using HLM in the presence and absence of reduced nicotinamide adenine dinucleotide phosphate (NADPH) cofactors were undertaken (Figure S2 in the Supporting Information). The major metabolite of 14 was a styrene derivative from hydroxylation at the benzylic carbon followed by elimination. With this information in hand, we replaced the 4-ethyl group in 14 with the metabolically blocked 4-OCF3 group in 16 with the hope of reducing intrinsic clearance. While this change led to increased potency, there was no improvement in intrinsic clearance. A metabolite identification study of 16 showed changes corresponding to oxidation of the tetrahydropyrido[4,3-d]pyrimidine core; several hydroxylated metabolites (and peaks corresponding to elimination of water from these species) were found.26 From these experiments, we concluded that there is no single metabolic soft spot to block within this series and that reduction in clearance would most likely be achieved through modulation of overall lipophilicity. However, our analogue program had demonstrated the challenges of achieving TGR5 potency at very low Log D.
The combination of potency, HLM clearance, and inability to block potential metabolic liabilities pointed to the selection of a compound that offered the best balance of attributes as a means to progress to in vivo studies. The best compounds at the extremes, that is, the most potent or the most metabolically stable, were assessed in DLM. While weaker in potency, compound 20 was selected on the basis of its lower HLM clearance. Compounds 16 and 18 were selected for further evaluation because of their potency. The in vitro human and dog microsomal intrinsic clearance are captured in Table 2. These compounds had moderate to high clearance in HLM and somewhat lower clearance in DLM.
On the basis of the balanced in vitro profile (uninduced EC50, DLM, and Log D), compound 16 was further assessed in an advanced battery of in vitro assays as well as in vivo dog pharmacokinetics study (Table 4). Of note was the high selectivity over CYP enzymes, selectivity over a wide panel of enzymes, receptors, and ion channels, and lack of activity against the farnasoid X receptor (FXR). In the dog pharmacokinetics study, 16 exhibited moderate clearance, low volume of distribution, short half-life, and good bioavailability.
Table 4. Pharmacological, ADME, and Safety Properties of 16.
| MW | 437.4 |
| cLog P | 2.30 |
| shake flask Log D | 3.03 |
| TPSA (Å2) | 110 |
| microsomal intrinsic CLint (μL/min/mg) | |
| dog | 115 |
| human (fu, mic 0.52) | 184 |
| CYP IC50 values | |
| 1A2, 2C9, 2C19, 2D6, 3A4 | all >30 μM |
| CYP metabolism phenotypinga | |
| 2C19 | 5% |
| 2C9 | 30% |
| 3A4 | 65% |
| thermodynamic solubility | |
| pH 6.5, phosphate buffered saline, 12 μg/mL (27 μM) | |
| plasma protein binding (% free, fu) | |
| dog | 2.6 |
| human | 8.3 |
| permeability [Papp (cm/s)] | |
| RRCK | AB 14.7 × 10–6 |
| MDR1-MDCK | AB 12.4 × 10–6 |
| BA 19.2 × 10–6 | |
| Genetox | Ames assay negative |
| hERGb inhibition (patch clamp) | IC50 = 4.6 μM |
| CEREP panel (82 assays) | IC50 > 10 μM |
| FXR (antagonist and agonist mode)27 | IC50 > 10 μM |
| in vivo pharmacokinetics | |
| CL (mL/min/kg) | dog, 8.05 |
| Vss (L/kg) | 0.43 |
| t1/2 (h) | 0.84 |
| F (%) | 41 |
Approximate percentage of drug-metabolizing isoform responsible for metabolism.
hERG = human ether-a-go-go-related gene.
On the basis of hTGR5 potency, its correlation to ex vivo assays linked to systemic TGR5 activation, dTGR5 potency, moderate in vivo dog clearance, and an acceptable in vitro safety profile, 16 was advanced to a 7 day toxicology study in dog. Of significant note, treatment-dependent changes in heart rate and blood pressure were noted for 16 as well as for another compound from a distinct and highly dissimilar chemotype,25 with no treatment-related changes to electrocardiogram waveform parameters. These data infer that a mechanism-based toxicity associated with systemic exposure of this compound is likely involved.
In conclusion, a potent and orally available TGR5 agonist 16 was identified starting from HTS hit 5. Compound 16 demonstrated an adequate dog in vivo pharmacokinetic profile that allowed for assessment of the effect of this systemically available TGR5 activator. A more detailed description of the in vitro and in vivo biological effects of 16 will be reported separately.
Acknowledgments
We thank Alex Gontcharov for the scale-up of 16 for toxicology studies, Jeffrey Dubins and Denise Gautreau for early work on assay development, and Daniel J. Lettiere for the toxicological study of 16 in dogs.
Glossary
Abbreviations
- ADME
absorption, distribution, metabolism, excretion
- cAMP
cyclic adenosine 5′-monophosphate
- CHO
Chinese hamster ovary
- CYP
cytochrome P450
- DLM
dog liver microsomes
- EDCI
1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
- FXR
farnasoid X receptor
- GLP-1
glucagon like peptide-1
- GPCR
G-protein-coupled receptor
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- hERG
human ether-a-go-go-related gene
- HLM
human liver microsomes
- HTS
high-throughput screening
- LPS
lipopolysaccharides
- MDCK Madin–Darby canine kidney
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- PBMC
peripheral blood mononuclear cells
- PEPPSI-IPr
pyridine-enhanced precatalyst preparation stabilization and initiation-isopropyl
- RLM
rat liver microsomes
- RRCK
Ralph Russ canine kidney
- SARs
structure–activity relationships
- TGR5
Takeda G-protein-coupled receptor 5
- TNF-α
tumor necrosis factor-α
- TPSA
topological polar surface area
Supporting Information Available
Experimental procedures, analytical data, and the description of in vitro and ex vivo assay systems. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Tiwari A.; Maiti P. TGR5: An Emerging Bile Acid G-protein Coupled Receptor Target for the Potential Treatment of Metabolic Disorders. Drug Discovery Today 2009, 14, 523–530. [DOI] [PubMed] [Google Scholar]
- Zhong M. TGR5 as a Therapeutic Target for Treating Obesity. Curr. Top. Med. Chem. 2010, 10, 386–396. [DOI] [PubMed] [Google Scholar]
- Pols T. W.; Noriega L. G.; Nomura M.; Auwerx J.; Schoonjans K. The Bile Acid Membrane Receptor TGR5 as an Emerging Target in Metabolism and Inflammation. J. Hepatol. 2011, 54, 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. Bile Acid Receptor Modulators in Metabolic Diseases. Annu. Rep. Med. Chem. 2011, 46, 69–87. [Google Scholar]
- Katsuma S.; Hirasawa A.; Tsujimoto G. Bile Acids Promote Glucagon-like Peptide-1 Secretion Through TGR5 in a Murine Enteroendocrine Cell Line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [DOI] [PubMed] [Google Scholar]
- Keitel V.; Reinehr R.; Gatsios P.; Rupprecht C.; Görg B.; Selbach O.; Häussinger D.; Kubitz R. The G-protein Coupled Bile Salt Receptor TGR5 is Expressed in Liver Sinusoidal Endothelial Cells. Hepatology 2007, 45, 695–704. [DOI] [PubMed] [Google Scholar]
- Pols T. W.; Nomura M.; Harach T.; Lo Sasso G.; Oosterveer M. H.; Thomas C.; Rizzo G.; Gioiello A.; Adorini L.; Pellicciari R.; Auwerx J.; Schoonjans K. TGR5 Activation Inhibits Atherosclerosis by Reducing Macrophage Inflammation and Lipid Loading. Cell. Metab. 2011, 14, 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-D.; Chen W.-D.; Yu D.; Forman B. M.; Huang W. The G-Protein-Coupled Bile Acid Receptor, Gpbar1 (TGR5), Negatively Regulates Hepatic Inflammatory Response Through Antagonizing Nuclear Factor Kappa Light-Chain Enhancer of Activated B Cells (NF-κB) in Mice. Hepatology 2011, 54, 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M.; Houten S. M.; Mataki C.; Christoffolete M. A.; Kim B. W.; Sato H.; Messaddeq N.; Harney J. W.; Ezaki O.; Kodama T.; Schoonjans K.; Bianco A. C.; Auwerx J. Bile Acids Induce Energy Expenditure by Promoting Intracellular Thyroid Hormone Activation. Nature 2006, 439, 484–489. [DOI] [PubMed] [Google Scholar]
- Brufau G.; Bahr M. J.; Staels B.; Claudel T.; Ockenga J.; Böker K. H. W.; Murphy E. J.; Prado K.; Stellaard F.; Manns M. P.; Kuipers F.; Tietge U. T. F. Plasma Bile Acids are not Associated with Energy Metabolism in Humans. Nutr. Metab. (London) 2010, 7, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H.; Macchiarulo A.; Thomas C.; Gioiello A.; Une M.; Hofmann A. F.; Saladin R.; Schoonjans K.; Pellicciari R.; Auwerx J. Novel Potent and Selective Bile Acid Derivatives as TGR5 Agonists: Biological Screening, Structure-Activity Relationships, and Molecular Modeling Studies. J. Med. Chem. 2008, 51, 1831–1841. [DOI] [PubMed] [Google Scholar]
- Pellicciari R.; Gioiello A.; Macchiarulo A.; Thomas C.; Rosatelli E.; Natalini B.; Sardella R.; Pruzanski M.; Roda A.; Pastorini E.; Schoonjans K.; Auwerx J. Discovery of 6α-Ethyl-23(S)-methylcholic Acid (S-EMCA, INT-777) as a Potent and Selective Agonist for the TGR5 Receptor, a Novel Target for Diabesity. J. Med. Chem. 2009, 52, 7958–7961. [DOI] [PubMed] [Google Scholar]
- Thomas C.; Gioiello A.; Noriega L.; Strehle A.; Oury J.; Rizzo G.; Macchiarulo A.; Yamamoto H.; Mataki C.; Pruzanski M.; Pellicciari R.; Auwerx J.; Schoonjans K. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell. Metab. 2009, 10, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans K. A.; Budzik B. W.; Ross S. A.; Wisnoski D. D.; Jin J.; Rivero R. A.; Vimal M.; Szewczyk G. R.; Jayawickreme C.; Moncol D. L.; Rimele T. J.; Armour S. L.; Weaver S. P.; Griffin R. J.; Tadepalli S. M.; M. R. Jeune M. R.; T. W. Shearer T. W.; Z. B. Chen Z. B.; Chen L.; Anderson D. L.; Becherer J. D.; De Los Frailes M.; Colilla F. J. Discovery of 3-Aryl-4-isoxazolecarboxamides as TGR5 Receptor Agonists. J. Med. Chem. 2009, 52, 7962–7965. [DOI] [PubMed] [Google Scholar]
- Herbert M. R.; Siegel D. L.; Staszewski L.; Cayanan C.; Banerjee U.; Dhamija S.; Anderson J.; Fan A.; Wang L.; Rix P.; Shiau A. K.; Rao T. S.; Noble S. A.; Heyman R. A.; Bischoff E.; Guha M.; Kabakibi A.; Pinkerton A. B. Synthesis and SAR of 2-Aryl-3-aminomethylquinolines as Agonists of the Bile Acid Receptor TGR5. Bioorg. Med. Chem. Lett. 2010, 20, 5718–5721. [DOI] [PubMed] [Google Scholar]
- Budzik B. W.; Evans K. A.; Wisnoski D. D.; Jin J.; Rivero R. A.; Szewczyk G. R.; Jayawickreme C.; Moncol D. L.; Yu H. Synthesis and Structure–Activity Relationships of a Series of 3-Aryl-4-isoxazolecarboxamides as a New Class of TGR5 Agonists. Bioorg. Med. Chem. Lett. 2010, 20, 1363–1367. [DOI] [PubMed] [Google Scholar]
- Kenakin T.Agonists: The Measurement of Affinity and Efficacy in Functional Assays. In A Pharmacology Primer: Theory, Applications, and Methods, 3rd ed.; Academic Press: New York, 2009; Chapter 5, pp 81–100. [Google Scholar]
- A comparison of reported TGR5 potency with values from our induced and uninduced assays is shown in the Supporting Information. Several literature assays are based on recombinant cells lines that appear to have TGR5 receptor levels higher than the native system. The reported potencies for selected agonists are generally aligned with our induced assay system.
- Di L.; Whitney-Pickett C.; Umland J. P.; Zhang H.; Zhang X.; Gebhard D. F.; Lai Y.; Federico J. J.; Davidson R. E.; Smith R.; Reyner E. L.; Lee C.; Feng B.; Rotter C.; Varma M. V.; Kempshall S.; Fenner K.; El-Kattan A. F.; Liston T. E.; Troutman M. D. Development of a New Permeability Assay Using Low-efflux MDCKII Cells. J. Pharm. Sci. 2011, 100, 4974–4985. [DOI] [PubMed] [Google Scholar]
- This lower expressing system provided a more conservative estimate of agonist potency and intrinsic activity. Detailed in vitro TGR5 pharmacology will be reported elsewhere.
- Valente C.; Belowich M. E.; Hadei N.; Organ M. G. Pd-PEPPSI Complexes and the Negishi Reaction. Eur. J. Org. Chem. 2010, 4343–4354. [Google Scholar]
- Please see the Supporting Information.
- Manteau B.; Genix P.; Brelot L.; Vors J.-P.; Pazenok S.; Giornal F.; Leuenberger C.; Leroux F. R. A General Approach to (Trifluoromethoxy)pyridines: First X-ray Structure Determinations and Quantum Chemistry Studies. Eur. J. Org. Chem. 2010, 6043–6066. [Google Scholar]
- Lombardo F.; Shalaeva M. Y.; K. A. Tupper K. A.; Gao F. ElogDoct: A Tool for Lipophilicity Determination in Drug Discovery. 2. Basic and Neutral Compounds. J. Med. Chem. 2001, 44, 2490–2497. [DOI] [PubMed] [Google Scholar]
- Futatsugi K.; Bahnck K. B.; Brenner M. B.; Buxton J.; Chin J. E.; Coffey S. B.; Dubins J.; Flynn D.; Gautreau D.; Guzman-Perez A.; Hadcock J. R.; Hepworth D.; Herr M.; Hinchey T.; Janssen A. M.; Jennings S. M.; Jiao W.; Lavergne S. Y.; Li B.; Li M.; Munchhof M. J.; Orr S. T. M.; Piotrowski D. W.; Roush N. S.; Sammons M.; Stevens B. D.; Storer G.; Wang J.; Warmus J. S.; Wei L.; Wolford. A. C.. Optimization of Triazole-based TGR5 Agonists Towards Orally Available Agents. Med. Chem. Commun., published online September 26, 2012; DOI: 10.1039/c2md20174g. [DOI] [Google Scholar]
- A subset of compounds that incorporated methyl, fluoro, or deutero substituents onto the core were designed to address metabolic soft spots. The substituted compounds that were assessed had lower potency and equivalent or higher clearance relative to that of 16. A full description of deuteration and metabolic fate of compounds related to 16 will be published in due course. For metabolic blocking in a related core, see the following:Nagle A.; Wu T.; Kuhen K.; Gagaring K.; Borboa R.; Francek C.; Chen Z.; Plouffe D.; Lin X.; Caldwell C.; Ek J.; Skolnik S.; Liu F.; Wang J.; Chang J.; Li C.; Liu B.; Hollenbeck T.; Tuntland T.; Isbell J.; Chuan T.; Alper P. B.; Fischli C.; Brun R.; Lakshminarayana S. B.; Rottmann M.; Diagana T. T.; Winzeler E. A.; Glynne R.; Tully D. C.; Chatterjee A. K. Imidazolopiperazines: Lead Optimization of the Second-Generation Antimalarial Agents. J. Med. Chem. 2012, 55, 4244–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assays conducted at Caliper Life Sciences (catalog #100-1051 and 100-1052).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.