

Abstract
Herein we describe the application of fragment-based drug design to bacterial DNA ligase. X-ray crystallography was used to guide structure-based optimization of a fragment-screening hit to give novel, nanomolar, AMP-competitive inhibitors. The lead compound 13 showed antibacterial activity across a range of pathogens. Data to demonstrate mode of action was provided using a strain of S. aureus, engineered to overexpress DNA ligase.
Keywords: Bacterial DNA ligase, S. aureus, fragment-based drug design, structure-based optimization
Bacterial DNA ligase (LigA) is an NAD+-dependent enzyme, which is essential for DNA replication and has attracted interest as a novel target for antibacterial therapy.1 LigA is responsible for ligating two strands of DNA via the formation of a phosphodiester bond between the 3′-hydroxyl end of one oligonucleotide and the 5′-phosphate end of another.2,3 This process involves a three-step mechanism. Initially, reaction between NAD+ and an active-site lysine leads to an adenylated form of the protein. Enzyme-bound AMP is then transferred to the 5′-phosphate end of a nicked DNA strand. Finally, attack on the AMP-DNA bond by the 3′-hydroxyl of a second strand of DNA seals the phosphate backbone and releases AMP. LigA has been shown to be essential for viability in all Gram-positive and Gram-negative organisms tested to date.4,5 It is highly conserved across bacterial species and is phylogenetically quite distinct from its human, ATP-dependent, counterpart. This provides encouragement that inhibitors of LigA may exhibit both broad-spectrum antibacterial activity and selectivity over human isozymes.6
Widespread bacterial resistance to current classes of approved antibiotics has led to an unmet medical need for compounds with novel modes of action that are not compromised by pre-existing resistance mechanisms.7 As a clinically unexploited target, inhibitors of LigA would fall into this category, and several classes of compounds have been reported in the literature.8−11 In most cases, high throughput screening (HTS) hits have provided chemical starting points, which were then optimized, often using structure-based drug design. Notable examples, shown to exhibit target-mediated antibacterial activity, include the adenosine analogue 1 and naphthyridine 2 (Figure 1).8,10
Figure 1.

Although in cases such as LigA molecular targeted HTS has delivered chemical leads, success rates from antibacterial HTS are typically lower than for targets from other therapeutic areas.12 In this letter, we describe an alternative approach to the discovery of LigA inhibitors using fragment-based drug design (FBDD).13 FBDD has become established as an approach for the generation of chemical leads for drug targets and has been employed in a variety of therapeutic areas, including antibacterials.14,15 In this approach specialized detection methods are used to identify small chemical compounds, fragments (MW ≤ 16 heavy atoms) that bind to the drug target, and structural biology is usually employed to establish their binding mode and to facilitate their optimization. A key feature of fragment-derived leads is that they are, on average, significantly smaller and less lipophilic than historical leads derived from HTS.16 Given that successful antibiotics are typically associated with lower lipophilicity than other drug classes,17,18 FBDD seems well suited to this therapeutic area. To our knowledge, this is the first published example of a fragment-derived LigA inhibitor.
An essential prerequisite to a successful fragment screening cascade for LigA was the identification of a stable, deadenylated, soakable crystal form of the protein. Constructs from multiple pathogens, including E. faecalis, H. influenzae, S. aureus, and E. coli, were prepared and subject to deadenylation by incubation with nicotinamide mononucleotide (NMN). LigA constructs showing reproducible and sustained levels of complete deadenylation (as determined by mass spectrometry) were progressesd to large scale crystallization trials. Of these, LigA from S. aureus provided the most stable soakable crystal form of the enzyme, and this was selected as the preferred system for X-ray crystallographic screening. Approximately 1500 compounds from our fragment library were then screened using a combination of high throughput LigA X-ray crystallography, ligand observed NMR (via water LOGSY), and a thermal shift (Tm) assay. Hits were then followed up using isothermal titration calorimetry (ITC) to determine binding affinities and ligand efficiencies (LE).19
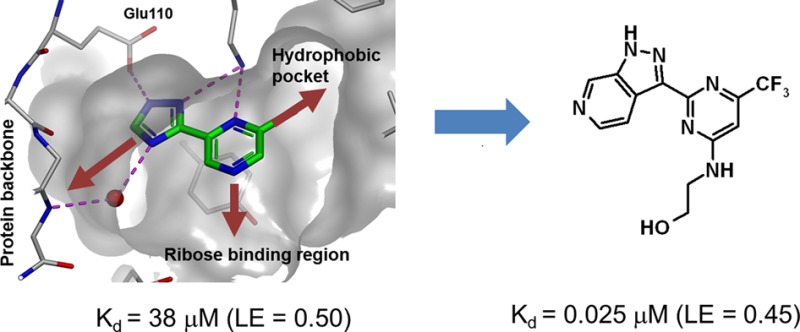
The starting point for chemical optimization was provided by the fragment-based screening hit 3 [Figure 2, Kd (ITC) = 38 μM, ligand efficiency, LE = 0.50]. The X-ray crystallographic structure of LigA (S. aureus) in complex with 3 revealed that the fragment binds in the AMP pocket and forms hydrogen bonds with the side chains of Lys283 and Glu110 (Figure 3a). An additional, water-mediated, hydrogen bond exists between the triazole nitrogen and the protein backbone NH of Ile113. Further interaction is provided by the side chain of Tyr219, which partially stacks with the pyrazine ring.
Figure 2.

Fragment optimization and growth of 3 toward 13. Data presented as Kd (ITC) or IC50 (biochemical assay) using DNA ligase from S. aureus.
Figure 3.
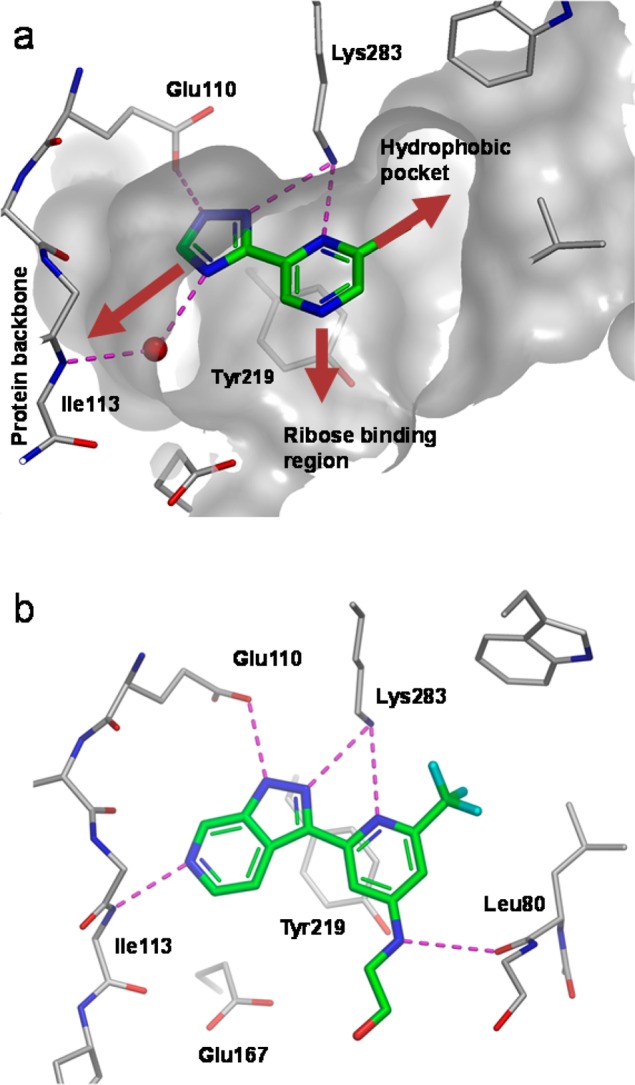
(a) X-ray crystal structure of fragment 3 bound to LigA (S. aureus) showing key hydrogen bonds (purple dotted lines), a key water molecule (red sphere), and partially resected Connoly surface. (gray). Growth vectors toward the hydrophobic pocket, ribose binding region, and the protein backbone are shown by the red arrows. (b) X-ray crystal structure of 12 bound to LigA (S. aureus).
Pyrazine 3 offered an attractive starting point for structure-based optimization for three reasons. First, 3 was one of the most ligand efficient hits observed for LigA. Second, the 2-chloro group partly fills the hydrophobic pocket (Figure 3a), a region of the active site which is occluded in the human form of the enzyme, providing an opportunity to obtain selectivity.20 Third, replacing the pyrazine with a pyridine would provide a growth vector to explore the ribose-binding region, via the pyridyl 4-position, provided this replacement was tolerated.
The pyridine analogue 4 [Kd (ITC) = 10 μM, IC50 = 17 μM, LE = 0.54] (Figure 2) was prepared, which, gratifyingly, showed improved activity over the fragment hit 3. One concern was the potentially reactive chlorine at position 2. To mitigate this risk, a number of alternative groups were explored at this position. Deletion of the 2-substituent (compound 5) or introduction of an electron donating substituent (e.g., OMe analogue 6) were both detrimental to activity. 2-Alkyl substituents (e.g., cyclobutyl 8), although appearing to fill the hydrophobic region more effectively than chlorine, surprisingly offered little advantage. These findings suggested that the electronics of the 2-substituent may be more important than sterics and that increasing the electron withdrawing nature of the alkyl group could be advantageous. The trifluoromethyl analogue 10 (IC50 = 16 μM, LE = 0.44) was synthesized, which proved equipotent with 4, albeit with a modest loss in ligand efficiency.
As described above, one of the interactions formed by these compounds is a water-mediated hydrogen bond between the triazole 4-position nitrogen and the backbone NH of Ile 113 (shown in Figure 3a). Our next strategy was to expand the triazole ring, in order to fill the pocket more effectively, displace this bridging water molecule and form a direct hydrogen bond with Ile 113.
To this end, the 6-azaindazole analogue 11 (IC50 = 0.22 μM, LE = 0.48) was prepared, which proved to be ∼70-fold more potent than 10. An X-ray structure of 11 could not be obtained, which we ascribed to the low solubility (solubility = 12 μg/mL)21 of this compound. Next, attention was turned toward exploring substitution at the pyridine 4-position in order to access the ribose-binding pocket. The more soluble ethanolamine derivative 12 (solubility = 113 μg/mL) was prepared, which was comparable in potency to 11 and enabled an X-ray structure of the protein–ligand complex to be obtained. The structure of 12 complexed with DNA ligase clearly shows N6 of the 6-azaindazole making the intended hydrogen-bond to the backbone NH of Ile113 (Figure 3b). The other interactions are conserved. An inspection of the ribose-binding region indicates a hydrogen-bond between the backbone carbonyl of Leu80 and the 4-aminopyridine substituent. However, the electron density around the last two atoms of the ethanolamine chain was ambiguous, and therefore, the exact position of the OH group is uncertain. Although the ethanolamine motif did not improve potency in the biochemical assay, it did result in 12 being the first analogue to show signs of antibacterial activity, particularly against S. aureus [MIC S.aureus (Oxford) = 8 μg/mL] (Table 1).
Table 1. MIC Data for Compounds 1, 12, and 13.
| MIC
(μg/mL) |
|||
|---|---|---|---|
| compd | 1a | 12 | 13 |
| Gram-Positive Pathogens | |||
| S. aureus Oxford | 8 | 8 | 4 |
| S. pneumoniae 1629 | 2 | 64 | 2 |
| S. pyogenes 1307006P | 8 | 128 | 8 |
| Gram-Negative Pathogens | |||
| H. influenzae H128 | 16 | 128 | 16 |
| E. coli 7623 | >128 | >128 | >128 |
| K. pneumonia 1161486 | >128 | >128 | >128 |
| P. aeruginosa PAO(MV) | >128 | >128 | >128 |
| A. baumannii BM4454 | >128 | >128 | >128 |
| Gram-Negative Efflux Knockout Strainsb | |||
| H. Influenzae H128 Acr B- | 2 | 8 | 1 |
| E. coli 7623 TolC- | 8 | 128 | 16 |
| K. pneumonia 1161486a TolC- | 8 | >128 | 32 |
| P. aeruginosa PAO322 | 64 | >128 | 32 |
| A. baumannii BM4652 (efflux mutant) | NT | >128 | 32 |
| cytotox IC50 (μg/mL) | NT | 16 | >36 |
Compound described in ref (8).
Engineered bacterial strains lacking key efflux transporter proteins (e.g., AcrB, TolC).
Further analysis of the binding mode led us to the hypothesis that compound 12 almost certainly binds in an unfavorable conformation.22 This is due to both steric (hydrogens) and electrostatic (heterocyclic nitrogen lone pairs) clashes between the pyridine and azaindazole rings (Figure 4). This is not the case in the more favored (unbound) conformation. Replacing the pyridine with a pyrimidine would remove the steric hindrance in the bound conformation and also create a clash (between nitrogen lone pairs) in the unbound conformation. Overall, this would contribute to stabilization of the bound state and therefore, hopefully, an increase in potency. Consequently, pyrimidine 13 [Kd (ITC) = 0.025 μM, LE = 0.45] (Figure 2) was prepared, which proved to be 15-fold more potent (compared to 12).
Figure 4.
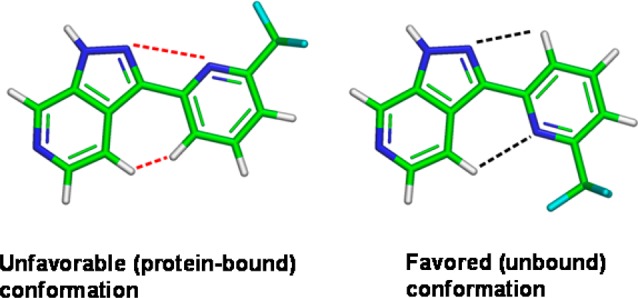
Illustration showing the alternative conformations of 12 (ethanolamine side chain removed for clarity).22 Electrostatic and steric clashes are shown in red, and positive interactions are shown in black.
Compound 13 exhibited antibacterial activities (Table 1), with MIC values of 8 μg/mL or less against wild-type Gram-positive pathogens S. aureus, S. pneumoniae, and S. pyogenes. Moderate activities (16–32 μg/mL) were also seen against efflux mutants of Gram-negative pathogens such as E. coli, P. aeruginosa, and A. baumannii. The lack of activity against most wild-type Gram-negative strains suggested that compound 13 was subject to active efflux out of the cell. This may also be compounded with inadequate cell permeability. General cytotoxicity against a mouse lung lymphoma cell line was not observed for compound 13 up to the highest concentration tested (100 μM). MIC data for literature compound 1 is shown for comparison.8
Antibacterial activity was determined to be caused by inhibition of LigA using a strain of S. aureus engineered to overexpress DNA ligase (Table 2). MICs of compound 13 were found to correlate with expression levels of DNA ligase.
Table 2. MIC for 13 in S. aureus Parent and LigA Overexpression Strainsa.
| MIC
(μg/mL) vs S. aureus RN4220 |
||||
|---|---|---|---|---|
| pYH4 |
pYH4-YerG
(DNA ligase) |
|||
| compd | – | + | – | + |
| 13 | 8 | 8 | 32/16 | 128 |
pYH4 = vector control. pYH4-YerG = vector + DNA gene. +/– = with or without inducer (0.1 μg/mL anhydrotetracycline).
This result is consistent with a DNA-ligase mediated mode of antibacterial action in this species. In the Gram-negative pathogen E. coli, the mode of action was also tracked to LigA inhibition by showing cross-resistance with LigA target mutants (Table 3).23
Table 3. MIC for 13 in E. coli Efflux Mutant Parent and LigA Target Mutant Strains.
| MIC (μg/mL) | |
|---|---|
| E. coli | compd 13 |
| TOP10 ΔTolC | 8 |
| TOP10 ΔTolC LigA G180E | 128/64 |
| TOP10 ΔTolC LigA R150S | 128/64 |
| TOP10 ΔTolC LigA R518H | 64 |
| E. coli LigA IC50 (uM) | 0.097 |
In summary, we have identified 13 (Kd = 25 nM, LE = 0.45, LLEAT = 0.4524) as an inhibitor of bacterial DNA ligase. Starting from pyrazine fragment 3 (Kd = 38 μM, LE = 0.50, LLEAT = 0.56), X-ray crystallographic data was used to establish key determinants for affinity and to guide structure based design. In particular, a strategy of establishing additional hydrogen bonds to the protein backbone and stabilizing the enzyme-bound conformation led to over a 1000-fold increase in activity, relative to starting point 3. Good LE and lipophilic ligand efficiency (LLEAT) were maintained during this process. Compound 13 demonstrated single-digit MICs across a range of Gram-positive pathogens, which, in the case of S. aureus, was shown to be target mediated. The 6-azaindaole scaffold provides a novel, nonpurine, chemotype to the LigA field, and to our knowledge, 13 is the first published example of a fragment-derived LigA inhibitor. Moreover, this work provides further validation of FBDD as an effective, complementary approach in the field of antibacterials.
Acknowledgments
We would like to thank David Rees, Andrew Leach, and Richard Jarvest for useful comments on the manuscript; and Joe Coyle, Finn Holding, Alex Thomas, Sharna Rich, Alan Rendina, Miriam Burman, and Emma Jones for biophysics and assay support. Also, thanks to Nicola Wallis, Christopher Johnson, Glyn Williams, Jeff Yon, and Christopher Murray for providing their support during the project.
Supporting Information Available
Assay conditions (including error limits and data for reference compounds), microbiology methods, biophysical methods, and synthetic procedure/characterization of compounds 4–13. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Shuman S. J. DNA ligases: Progress and prospects. J. Biol. Chem. 2009, 284, 17365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman I. R. DNA ligase: Structure, mechanism and function. Science 1974, 186, 790. [DOI] [PubMed] [Google Scholar]
- Tomkinson A. E.; Vijayakumar S.; Pascal J. M.; Ellenberger T. DNA ligase: Structure, reaction mechanism and function. Chem. Rev. 2006, 106, 687. [DOI] [PubMed] [Google Scholar]
- Streker K.; Schäfer T.; Freiberg C.; Brötz-Oesterhelt H.; Hacker J.; Labischinski H.; Ohlsen K. In vitro and in vivo validation of ligA and tarI as essential targets in Staphylococcus aureus. Antimicrob. Agents Chemother. 2008, 52, 4470–4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavesa-Curto M.; Sayer H.; Bullard D.; MacDonald A.; Wilkinson A.; Smith A.; Bowater L.; Hemmings A.; Bowater R. P. Characterisation of a temperature-sensitive DNA ligase from Escherichia coli. Microbiology 2004, 150, 4171–4180. [DOI] [PubMed] [Google Scholar]
- Swift R. V.; Amaro R. M. Discovery and design of DNA and RNA ligase inhibitors in infectious microorganisms. Expert Opin. Drug Discovry 2009, 4, 1281–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1. [DOI] [PubMed] [Google Scholar]
- Mills S. D.; Eakin A. E.; Buurman E. T.; Newman J. V.; Gao N.; Huynh H.; Johnson K. D.; Lahiri S.; Shapiro A. B.; Walkup G. K.; Yang W.; Stokes S. S. Novel bacterial NAD+-dependent DNA ligase inhibitors with broad-spectrum activity and antibacterial efficacy in vivo. Antimicrob. Agents Chemother. 2011, 1088, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes S. S.; Huynh H.; Gowravaram M.; Albert R.; Cavero-Tomas M.; Chen B.; Harang J.; Loch J. T. III; Lu M.; Mullen G. B.; Zhao S.; Liu C. F.; Mills S. D. Discovery of bacterial NAD+-dependent DNA ligase inhibitors: optimization of antibacterial activity. Bioorg. Med. Chem. Lett. 2011, 21, 4556. [DOI] [PubMed] [Google Scholar]
- Surivet J.-P.; Lange R.; Hubschwerlen C.; Keck W.; Specklin J.-L.; Ritz D.; Bur D.; Locher H.; Seiler P.; Strasser D. S.; Prade L.; Kohl C.; Schmitt C.; Chapoux G.; Ilhan E.; Ekambaram N.; Athanasiou A.; Knezevic A.; Sabato D.; Chambovey A.; Gaertner M.; Enderlin M.; Boehme M.; Sippel V.; Wyss P. Structure-guided design, synthesis and biological evaluation of novel DNA ligase inhibitors with in vitro and in vivo anti-staphylococcal activity. Bioorg. Med. Chem. Lett. 2012, 22, 6705–6711. [DOI] [PubMed] [Google Scholar]
- Wang T.; Duncan L.; Gu W.; O’Dowd H.; Wei Y.; Perola E.; Parsons J.; Gross C. H.; Moody C. S.; Arends R. S. J.; Charifson P. S. Design, synthesis and biological evaluation of potent NAD+-dependent DNA ligase inhibitors as potential antibacterial agents. Part II: 4-amino-pyrido[2,3-d]pyrimidin-5(8H)-ones. Bioorg. Med. Chem. Lett. 2012, 22, 3699–3703. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6129–40. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Rees D. C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [DOI] [PubMed] [Google Scholar]
- Eakin A. E.; Green O.; Hales N.; Walkup G. K.; Bist S.; Singh A.; Mullen G.; Bryant J.; Embrey K.; Gao N.; Breeze A.; Timms D.; Andrews B.; Uria-Nickelsen M.; Demeritt J.; Loch J. T. III; Hull K.; Blodgett A.; Illingworth R. N.; Prince B.; Boriack-Sjodin P. A.; Hauck S.; MacPherson L. J.; Ni H.; Sherer B. Pyrrolamide DNA gyrase inhibitors: Fragment-based NMR screening to antibacterial agents. Antimicrob. Agents Chemother. 2012, 5631240–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson S. A.; McLean K. J.; Surade S.; Yang Y. Q.; Leys D.; Ciulli A.; Munro A. W.; Abell C. Application of fragment screening and merging to the discovery of inhibitors of the mycobacterium tuberculosis cytochrome P450 CYP121. Angew. Chem., Int. Ed. 2012, 51, 9311–9316. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Verdonk M. L.; Rees D. C. Experiences of fragment-based drug discovery. Trends Pharmacol. Sci. 2012, 335224–232. [DOI] [PubMed] [Google Scholar]
- O’Shea R.; Moser H. E. Physicochemical properties of antibacterial compounds: implications for drug discovery. J. Med. Chem. 2008, 51, 2871–2878. [DOI] [PubMed] [Google Scholar]
- Manchester J. I.; Buurman E. T.; Bisacchi G. S.; McLaughlin R. E. Molecular determinants of AcrB-mediated bacterial efflux implications for drug discovery. J. Med. Chem. 2012, 55, 2532–2537. [DOI] [PubMed] [Google Scholar]
- LE = −ΔG/HAC ≈ −RT ln(IC50)/HAC.Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Han S.; Chang J. S.; Griffor M. Structure of the adenylation domain of NAD+-dependent DNA ligase from Staphylococcus aureus. Acta Crystallogr. 2009, F65, 1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqueous solubility measured by a HPLC method using ChemiLuminescent Nitrogen Detection quantification.
- Molecular calculations and NMR data to support this, together with precedence from the CCD, are described in the Supporting Information.
- Target mutants were generated using compound 1 (see Supporting Information).
- Mortenson P. N.; Murray C. W. J. Comput. Aided Mol. Des. 2011, 25, 663–667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.