

Abstract
Recent observations on the emergence of artemisinin resistant parasites have highlighted the need for new antimalarial treatments. An HTS campaign led to the identification of the 1-(1-aminopropan-2-ol)carbazole analogues as potent hits against Plasmodium falciparum K1 strain. The SAR study and optimization of early ADME and physicochemical properties direct us to the selection of a late lead compound that shows good efficacy when orally administrated in the in vivo P. berghei mouse model.
Keywords: Malaria, WHO, SAR, carbazole, Plasmodium falciparum, Plasmodium berghei, IC50, hERG
One of the most mortal parasitic diseases remains malaria, with an estimated one million deaths per year.1 Even if Plasmodium species, the parasite responsible for malaria, often infects humans, the Plasmodium falciparum strains are responsible for most of the death caused by the disease.2 The main affected populations are children under five years old and pregnant women.3 Dramatically one child dies from malaria every 30 s.1,4
The extensive use of well-known antimalarial drugs led to the emergence of resistance of certain strains to the current marketed drugs (e.g., chloroquine, Figure 1)5 urging the international community to develop new chemotherapeutic agents. The discovery of a highly active compound (artemisinin, Figure 1)6 led to the development of several endoperoxide analogues, used mainly within combination therapies.7 Despite this precaution, recent studies have reported a possible emergence of resistance against this drug and its derivatives.8−10 This worrying observation emphasizes the importance of research for new drugs against malaria.4,8−14
Figure 1.

Structure and IC50 values of antimalarials against Pf-K1.12−16
As the identification and validation of new targets remains challenging,17 most research programs use known antimalarials for pharmacophores11 and rely on phenotypic screening.18 Importantly the drug candidate should be efficacious against a large panel of Plasmodium species and show activity in a preclinical murine model. It should also be safe and stable in extreme conditions and require low cost of goods.1
The World Health Organisation (WHO) supported several programs to identify new chemical entities for the fight against malaria. Among several thousands of compounds screened within a public-private partnership collaboration with Tropical Diseases Research (TDR)–WHO,19 the commercially available compound TDR30137 related to N-substituted carbazoles has emerged as a hit in a phenotypic in vitro screen against the chloroquine-resistant P. falciparum K1 (Pf-K1) in human red blood cells with an IC50 of 57 nM (Figure 1). Although the hit exhibited good in vitro potency, it did not show any activity in the in vivo P. Berghei mouse model used. Interestingly, carbazole scaffolds are found in nature with reported antimalarial activity.20,21 Furthermore, synthetic carbazoles were also described as potential antimalarial agents,22 Bax channel modulators,23 and neuroprotective agents.24,25 Thus, we thought that TDR30137 was a good starting point for a medicinal chemistry program. Herein, we report the stucture–activity relationship (SAR) study, the physicochemical and in vitro and in vivo DMPK profiling, the in vivo efficacy, and finally the preliminary safety profile of a selected series of compounds that led quickly to the identification of an antimalarial lead.
In an effort to extend the knowledge around the new antimalarial hit TDR30137 (Figure 1), substructure searches and the synthesis of new analogues was performed to review the importance of the carbazole, the aromatic moiety, the spacer, and the amine type. Eighty compounds were selected, using substructure search, from Merck Serono screening library. Many modifications led to inactive compounds. Key potent features were found in the substituted carbazole linked to an amine via a floppy chain. Furthermore, 40 analogues were synthetised to refine SAR.
Compounds were evaluated through a parasitic growth assay at Swiss Tropical and Public Health Institute (Swiss TPH). This assay is based on the measurement of the incorporation of hypoxanthine after 3 days of incubation of a mixture of the evaluated drug, human red blood cells, and P. falciparum. SAR studies were performed by profiling analogues highlighting significant changes in terms of structure and behavior of substituents used. IC50s were reported to compare and rank the differently substituted analogues (Table 1). Such compounds were obtained via a two to five step straightforward synthesis from carbazole 1 and other cheap and readily available starting materials (see all details in Supporting Information). Among the most representative compounds for SAR evaluation, analogues 12 to 15 are summarized in Table 1.26
Table 1. Structure–Activity Relationship Study.
| compd | R1 | R2 | R3 | Pf-K1 IC50a | cpKab |
|---|---|---|---|---|---|
| 12 | Br | Br | OH | 9 nM | 10.0 |
| 13a | Cl | Cl | OH | 16 nM | 9.4 |
| 13b | Br | Br | F | 106 nM | 9.4 |
| 13c | Cl | Cl | =O | 448 nM | 9.4 |
| 13d | Cl | Cl | H | 707 nM | 9.4 |
| 14a | Br | Br | OH | 27 nM | 7.5 |
| 14b | Cl | H | OH | 165 nM | 7.5 |
| 14c | H | H | OH | 275 nM | 7.5 |
| 14d | H | H | OMe | 3313 nM | 7.5 |
| 15 | Cl | Cl | OH | 344 nM | 5.3 |
A 72 h assay in the presence of serum albumin.
Calculated pKa predicted using pkcalc 4.1.0; CompuDrug, based on published work.27
The removal of one or two halogens on the carbazole moiety (14a vs 14b and 14c, Figure 1) decreased activity of one log unit. In addition, the hydroxyl group and more particularly his hydrogen bound donor character seemed to be key for activity. Indeed, methyl ether 14d vs compound 14c, ketone 13c, fluoro 13b, and alkyl 13d vs 13a showed significant loss of activity. Finally, the basic character of the amine in the southern part seemed to play an important role in the activity on Pf-K1. For example, introduction of nonbasic functionality such as an amide (15) reduced the activity of one log unit. Other amides show the same tendency (data not shown). On the contrary, compounds 12 and 13a, combining the presence of hydrogen bond donor group and optimized pKa, showed excellent activity in low nanomolar range against Pf-K1.
In terms of selectivity, both compounds 12 and 13a had an excellent in vitro potency against a panel of resistant Pf strains (K1, NF54, D6, W2, TM91C235, 7G8, and VIS).28 Considering those promising in vitro results, the in vivo efficacy of the two compounds was assessed in a P. Berghei mouse model of infection. As compared to HuSCID model, which uses humanized mouse, the cheaper P. Berghei mouse model will ensure the possibility to have a greater amount of compounds tested in vivo.
As the chosen in vivo mouse model uses a Plasmodium strain different from the one used in the in vitro assays, (P. berghei vs P. falciparum, respectively), a correlation study was initiated, which confirmed the validity of the in vivo P. Berghei mouse model.29
Both compounds 12 and 13a exhibited acceptable in vitro physicochemical and eADME characteristics for such lipophilic heterocyclic amines.29 Interestingly, the low to medium intrinsic clearance measured translated into a medium in vivo clearance in mouse after intravenous administration at 1 mg/kg (∼15% and ∼30% of liver blood flow for compounds 12 and 13a, respectively).29 The intravenous volume of distribution was high for both amino compounds (13 L/kg and 15 L/kg for compounds 12 and 13a, respectively) suggesting extensive distribution of the compound from the blood. Following oral administration, at 5 mg/kg, a favorable bioavailability of 50% was apparent, along with a long terminal half-life of 15 and 8 h for compounds 12 and 13a, respectively. Furthermore, both compounds showed high Cmax values (158 and 135 ng/mL for compounds 12 and 13a, respectively). Compound 12 showed a higher AUC than 13a (3753 h·ng/mL versus 1583), indicating a potential higher concentration in tissue as compared to compound 13a. The pharmacokinetic profile was linear between oral doses of 3 and 100 mg/kg for compound 13a, and between 3 and 30 mg/kg for compound 12. Following these encouraging results in terms of clearance and bioavailability, an in vivo experiment in a mouse model infected with the P. berghei strain of the parasite was performed for both compounds.
The in vivo activity was assessed in the P. berghei mouse model using a 4-day once daily dosing regimen of the test compounds. The animals were treated orally on days 0–3 starting 4 h postinfection using dose groups of 3, 10, 30, and 100 mg/kg/day. Red blood cell parasitemia and plasma exposure were determined on day 4, 24 h after the last dose, followed by the monitoring of mean survival days (MSD) of the mice during 26 additional days.33
As shown in Figure 2A, the parasitemia of P. berghei infected mice was reduced in a dose-dependent manner with ED50 values of 11.4 and 34.5 mg/kg/day for compounds 12 and 13a, respectively. By using the highest dose of 100 mg/kg/day, the total parasitemia could be reduced by 99.97% (12) and 99.93% (13a), therefore showing an almost complete disappearance of parasites in the mice after only 4 days of treatment.
Figure 2.
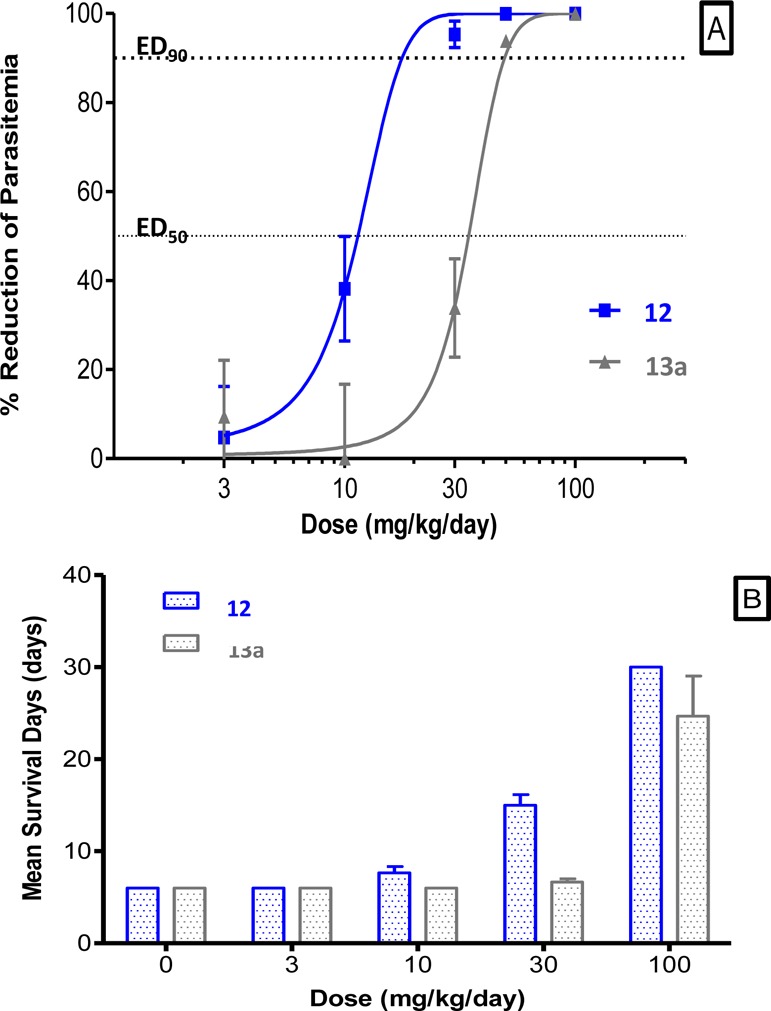
Dose dependency of activity of compounds 12 and 13a against Pf-K1. (A) Dose response curve of compounds 12 (blue) and 13a (gray) in the P. berghei infected mice (4 day-treatment, po). (B) Mean survival days (MSD) of P. berghei infected mice after 4-day treatment po.
In addition to the assessment of parasitemia reduction, the survival of the animals was also monitored during 26 additional days. Figure 2A shows that parasitemia was reduced by 38% after administration of 10 mg/kg/day of 12; nevertheless, as shown in Figure 2B, no effect on the mean survival of the mice was seen. However, an increase of the dose from 10 to 30 mg/kg/day of compound 12 led to a significant increase of the mean survival days (MSD) of the mice. Concerning compound 13a, a minimum dose of 100 mg/kg/day is needed to reach a real increase of MSD. This dose of 100 mg/kg/day led to the survival of the mice during 30 days by using compound 12 after oral administration.
These in vivo experiments led to the selection of compound 12 as being the most promising compound, considering the increase of the survival of the animals treated by 100 mg/kg/day, as compared to its analogue 13a.30
Safety profile is of extreme importance while considering the situation of such patients, living mainly in areas with limited medical supervision. First genotoxicity and mutageniticity assays were performed. Racemate 12 was found negative in micronucleus and AMES in vitro assays, including under metabolic conditions.
Then, in order to assess the cardiac toxicity of compound 12, the first safety assays performed were focusing on human ether-a-go-go related gene channel (hERG channel). The inhibition of this cardiac ion channel can lead to cardiotoxicity, which has been reported for several antimalarial compounds (e.g., Halofantrin, Chloroquine, Ki = 7.5 μM; Mefloquine, Ki = 1.9 μM).31 The activity of compound 12 toward hERG was high, showing an inhibition of the channel of 99% at 10 μM. It is noteworthy that compound 12 did not display any inhibition against any other cardiac ion channels profiled (hNaV1.5, KV4.3, hCaV1.2, hKV1.5, hHCN4, and hKir2.1), suggesting that only hERG has to be taken into account.33
As every biological target, this activity on the ion channel could be linked to the stereochemistry of the compounds tested. To assess if the toxicity risk was only linked to one of the two enantiomers, the enantiopure compounds were prepared in a 3-step synthesis.26
The profiling of both enantiomers of compound 12 showed that compounds 18a and its enantiomer 18b had the same activity against Pf-K1, hERG, and the panel of CYPs than the racemate 12.29 Thus, the profiling of the two enantiomers did not show any improvement of safety parameters as compared to the racemate 12. Those parameters and mainly hERG inhibition should be the identified liability to solve, thanks to medicinal chemistry design of new compounds. The design of novel compounds would hence be based on the knowledge of hERG pharmacophore to decrease the activity on the channel.
The described pharmacophore for hERG inhibition (Figure 3A) shows that compounds 12, 18a, and 18b contain chemical fragments that displayed physicochemical properties inhibiting the activity on the channel (Figure 3B).32,34
Figure 3.
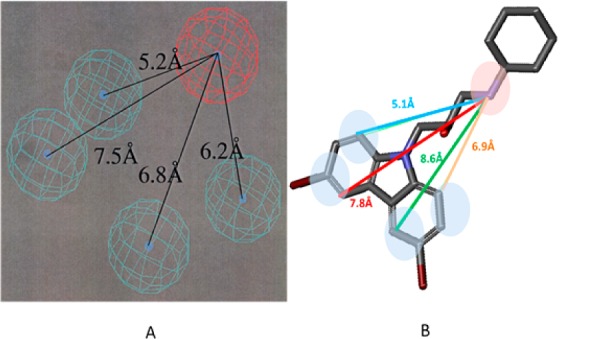
hERG pharmacophore vs compound 12 structure. (A) hERG pharmacophore as depicted in the literature.34 The blue spheres depict lipophilic areas and the red sphere represents an ionizable center. Reproduced with permission from ref (34). Copyright 2002 American Society for Pharmacology and Experimental Therapeutics. (B) Calculation of distances between the lipophilic centers (blue) and basic centers (red) of compound 12.
The distance between the lipophilic areas and ionic center of compounds 12, 18a, and 18b corresponds to the described pharmacophore, predicting a high affinity of the compounds to the ion channel.
Despite the great in vitro and in vivo activity of compounds 12, 18a, and 18b against the parasite, current work for optimization alleviating the hERG inhibition is evaluated.
Thanks to a Private Public Partnership between Merck Serono and WHO, a HTS campaign led to the identification of an interesting hit. The screening of about 80 analogues from Merck Serono screening library and a focused medicinal chemistry program helped to build a SAR, leading to the identification of compound 12, with in vivo efficacy. Nevertheless, safety evaluation revealed hERG inhibition, which could be challenging for the development toward a preclinical candidate. In parallel, a backup strategy, developing a different series in terms of structures and profiles, has been put in place relying on drastic changes of physicochemical properties35,36 of the series, which will be shortly communicated.
Acknowledgments
We thank Dr. B. Ramirez, Dr J.-M. Paris for fruitful conversations and helpful advices. We thank Dr. T. N. C. Wells, Dr. J. Burrows, and Dr. P. Willis for sharing their expertise during interesting conversations around antimalarial drug discovery processes. We thank Dr. W. Sauer for his input in building and confirming the correlation between Pf-K1 and P. berghei in vitro activities.
Glossary
Abbreviations
- CL
in vivo clearance
- CLint
intrinsic clearance
- CYP
cytochrome P450
- ED50
50% efficacy dose
- Fz
bioavailability
- hERG
human ether-a-go-go related gene
- HTS
high throughput screening
- IC50
50% inhibitory concentration
- IC90
90% inhibitory concentration
- MSD
mean survival days
- P. berghei
Plasmodium berghei
- Pf
Plasmodium falciparum
- PK
pharmacokinetic profile
- po
per os
- SAR
structure–activity relationship
- Vss
volume of distribution at steady state
- WHO
World Health Organization
Supporting Information Available
Synthesis and analytical data of intermediates and exemplary compounds. Details for Pf-K1 in vitro growth assay. P. berghei in vitro growth assay, microsomal stability, CYP inbibition in vitro assay, and rodent pharmacokinetic studies. P. berghei in vivo assay. hERG patchclamp assay. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ (J.M.) AC Immune SAPSE Building B, EPFLCH-1015 Lausanne, Switzerland.
Author Present Address
⊥ (H.B.) Simcyp Ltd. (a Certara Company), Blades Enterprise Center, Sheffield, S2 4SU, U.K.
Author Present Address
# (A.S.) Institut de Recherche Pierre Fabre, 17 Avenue Jean Moulin, 81106 Castres, France.
Author Present Address
○ (K.G.) MerckSerono, Alsfelder Straße 17, 64289 Darmstadt, DE, Germany.
Author Contributions
All authors have given approval to the final version of this manuscript.
This investigation was done for and received support from the UNICEF/UNDP/World Bank/WHO Special Program for Research and Training in Tropical Diseases (TDR).
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization. Malaria Fact Sheet, 2012. http://www.who.int/mediacentre/factsheets/fs094/en/print.html.
- Nosten F.; Rogerson S. J.; Beeson J. G.; McGready R.; Mutabingwa T. K.; Brabin B. Malaria in pregnancy and the endemicity spectrum: what can we learn?. Trends Parasitol. 2004, 20, 425–432. [DOI] [PubMed] [Google Scholar]
- Dev V.; Phookan S.; Sharma V. P.; Dash A. P.; Anand S. P. Malaria parasite burden and treat- ment seeking behaviour in ethnic communities of Assam, northeastern India. J. Infect. 2006, 52, 131–139. [DOI] [PubMed] [Google Scholar]
- Snow R. W.; Guerra C. A.; Noor A. M.; Myint H. Y.; Hay S. I. The global distribution of clinical episodes of plasmodium falciparum malaria. Nature 2005, 434, 214–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlemann A. C.; Krishna S. Antimalarial multi-drug resistance in Asia: mechanisms and assessment. Curr. Top. Microbiol. Immunol. 2005, 295, 39–53. [DOI] [PubMed] [Google Scholar]
- Afonso A.; Hunt P.; Cheesman S.; Alves A. C.; Cunha C. V.; do Rosário V.; Cravo P. Malaria Parasites Can Develop Stable Resistance to Artemisinin but Lack Mutations in Candidate Genes atp6 (Encoding the Sarcoplasmic and Endoplasmic Reticulum Ca2+ ATPase), tctp, mdr1, and cg10. Antimicrob. Agents Chemother. 2006, 50, 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman R. T.; Fidock D. A. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat. Rev. Microbiol. 2009, 7, 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noedl H.; Se Y.; Schaecher K.; Smith B. L.; Socheat D.; Fukuda M. M. Evidence of Artemisinin-Resistant Malaria in Western Cambodia. N. Engl. J. Med. 2008, 359, 2619–2620. [DOI] [PubMed] [Google Scholar]
- White N. J. Qinghaosu (Artemisinin): The price of success. Science 2008, 320, 330–334. [DOI] [PubMed] [Google Scholar]
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P.; Lindegardh N.; Socheat D.; White N. J. Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 2009, 361, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White N. J. Antimalarial drug resistance and combination chemotherapy. Philos. Trans. R. Soc., B. 1999, 354, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony M. P.; Burrows J. N.; Duparc S.; Jmoehrle J.; Wells T. N. C. The global pipeline of new medicines for the control and elimination of malaria. Malar. J. 2012, 11, 316–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olliaro P.; Well T. N. The global portfolio of new antimalarial medicines under development. Clin. Pharmacol. Ther. 2009, 85, 584. [DOI] [PubMed] [Google Scholar]
- Posner G. H.; Chang W.; Hess L.; Woodard L.; Sinishtaj S.; Usera A. R.; Maio W.; Rosenthal A. S.; Kalinda A. S.; D’Angelo J. G.; Petersen K. S.; Stohler R.; Chollet J.; Santo-Tomas J.; Snyder C.; Rottmann M.; Wittlin S.; Brun R.; Shapiro T. A. Malaria-infected mice are cured by oral administration of new artemisinin derivatives. J. Med. Chem. 2008, 51, 1035–1042. [DOI] [PubMed] [Google Scholar]
- Fügi M. A.; Wittlin S.; Dong Y.; Vennerstrom J. L. Antimicrob. Agents Chemother. 2010, 54, 1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramazani A.; Zakeri S.; Sardari S.; Khodakarim N.; Djadidt N. D. In vitro and in vivo anti-malarial activity of Boerhavia elegans and Solanum surattense. Malar. J. 2010, 9, 124–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler E. A. Malaria research in the post-genomic era. Nature 2008, 455, 751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L. M.; Jiménez-Díaz M. B.; Crespo B.; De-Cozar C.; Almela M. J.; Angulo-Barturen I.; Castañeda P.; Ibañez J.; Fernández E. P.; Ferrer S.; Herreros E.; Lozano S.; Martínez M. S.; Rueda L.; Burrows J. N.; García-Bustos J. F.; Gamo F. J. Cyclopropyl Carboxamides, a Chemically Novel Class of Antimalarial Agents Identified in a Phenotypic Screen. Antimicrob. Agents Chemother. 2011, 55, 5740–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaka S.; Hudson A. Innovative lead discovery strategies for tropical diseases. Nat. Rev. Drug Discovery 2006, 5, 941–955. [DOI] [PubMed] [Google Scholar]
- Thongthoom T.; Songsiang U.; Phaosiri C.; Yenjai C. Biological activity of chemical constituents from Clausena harmandiana. Arch. Pharm. Res. 2010, 33, 675–680. [DOI] [PubMed] [Google Scholar]
- Kongkathip N.; Kongkathip B. Constituents and bioactivities of Clausena excavata. Heterocycles 2009, 79, 121–144. [Google Scholar]
- DeGraw J. I.; Brown V. H.; Keyanpour-Rad M. 3- and 4-Carbazole dialkylaminocarbinols as potential antimalarial agents. J. Med. Chem. 1971, 14, 549–550. [DOI] [PubMed] [Google Scholar]
- Bombrun A.; Gerber P.; Casi G.; Terradillos O.; Antonsson B.; Halazy S. 3,6-Dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via bax channel modulation. J. Med. Chem. 2003, 46, 4365–4368. [DOI] [PubMed] [Google Scholar]
- MacMillan K. S.; Naido J.; Liang J.; Melito L.; Williams N. S.; Morlock L.; Huntington P. J.; Estill S. J.; longgood J.; Becker G. L.; McKnight S. L.; Pieper A. A.; De Brabander J. K.; Ready J. M. Development of proneurogenic, neuroprotective small molecules. J. Am. Chem. Soc. 2011, 133, 1428–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper A. A.; Xie S.; Capota E.; Estill S. J.; Zhong J.; Long J. M.; Becker G. L.; Huntington P.; Goldman S. E.; Shen C.-H.; Capota M.; Britt J. K.; Kotti T.; Ure K.; Brat D. J.; Williams N. S.; MacMillan K. S.; Naidoo J.; Melito L.; Hsieh J.; De Brabander J.; Ready J. M.; McKnight S. L. Discovery of a proneurogenic, neuroprotective chemical. Cell 2010, 142, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- General schemes for the synthesis of compounds 12 to 15 are depicted and developed in the Supporting Information.
- Csizmadia F.; Szegezdi J.; Darvas F.. Proceedings of the 9th European Symposium on Structure-Activity Relationships: QSAR and Molecular Modeling, September 7–11, 1992, Strasbourg, France; Wermuth C. G., Ed. ESCOM:Leiden, The Netherlands, 1993; pp 507–510. [Google Scholar]
- Data are detailed in the Supporting Information, in the paragraph “In vitro anti malarial activity (IC50 and IC90) for (12) against a panel of Plasmodium falciparum strains.”
- See Supporting Information in additional information chapter.
- For in vivo activities of related antimalarial drugs, please refer toCharman S. A.; Arbe-Barnes S.; Bathurst I. C.; Brun R.; Campbell M.; Charman W. N.; Chiu F. C. K.; Chollet J.; Craft J. C.; Creek D. J.; Dong Y.; Matile H.; Maurer M.; Morizzi J.; Nguyen T.; Papastogiannidis P.; Scheurer C.; Shackleford D. M.; Sriraghavan K.; Stingelin L.; Tang Y.; Urwyler H.; Wang X.; White K. L.; Wittlin S.; Zhou L.; Vennerstrom J. L. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc. Natl. Acad. Sci.U.S.A. 2011, 108, 4400–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traebert M.; Dumotier B.; Meister L.; Hoffmann P.; Dominguez-Estevez M.; Suter W. Inhibition of hERG K+ currents by antimalarial drugs in stably transfected HEK293 cells. Eur. J. Pharmacol. 2004, 484, 41–48. [DOI] [PubMed] [Google Scholar]
- Tobita M.; Nishikawa T.; Nagashima R. A discriminant model constructed by the support vector machine method for HERG potassium channel inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 2886–2890. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for experimental details.
- Ekins S.; Crumb W. J.; Sarazan R. D.; Wikel J. H.; Wrighton S. A. Three-dimensional quantitative structure–activity relationship for inhibition of human ether-a-go-go-related gene potassium channel. J. Pharmacol. Exp. Ther. 2002, 301, 427–434. [DOI] [PubMed] [Google Scholar]
- Jamieson C.; Moir E. M.; Rankovic Z.; Wishart G. Medicinal chemistry of hERG optimizations: Highlights and hang-ups. J. Med. Chem. 2006, 49, 5029. [DOI] [PubMed] [Google Scholar]
- Morgenthaler M.; Schweizer E.; Hoffmann-Röder A.; Benini F.; Martin R. E.; Jaeschke G.; Wagner B.; Fischer H.; Bendels S.; Zimmerli D.; Schneider J.; Diederich F.; Kansy M.; Müller K. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amine Basicities. ChemMedChem 2007, 2, 1100–1115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.