

Abstract
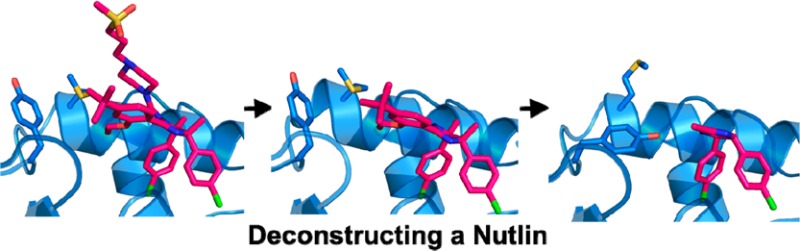
Protein–protein interaction (PPI) systems represent a rich potential source of targets for drug discovery, but historically have proven to be difficult, particularly in the lead identification stage. Application of the fragment-based approach may help toward success with this target class. To provide an example toward understanding the potential issues associated with such an application, we have deconstructed one of the best established protein–protein inhibitors, the Nutlin series that inhibits the interaction between MDM2 and p53, into fragments, and surveyed the resulting binding properties using heteronuclear single quantum coherence nuclear magnetic resonance (HSQC NMR), surface plasmon resonance (SPR), and X-ray crystallography. We report the relative contributions toward binding affinity for each of the key substituents of the Nutlin molecule and show that this series could hypothetically have been discovered via a fragment approach. We find that the smallest fragment of Nutlin that retains binding accesses two subpockets of MDM2 and has a molecular weight at the high end of the range that normally defines fragments.
Keywords: Nutlin, protein−protein interaction inhibitor, p53, MDM2, binding affinity
Inhibiting protein–protein interactions (PPI) with small molecules is a difficult objective but could potentially lead to a wide variety of novel and important therapeutics.1,2 There are several possible pathways toward the discovery of such molecules, including high-throughput screening of large compound libraries to obtain initial leads. Traditionally, these libraries have consisted of compounds in the molecular weight range 200–500 Da. More recently, a strategy employing libraries comprised only of small compounds, the fragment-based approach,3,4 has been gaining popularity. Practitioners have settled on a similar set of characteristics for the fragments comprising their libraries, with a molecular weight range established at 100–300 Da. However, protein–protein interaction systems represent a unique class of drug target, and it has already been shown that successful inhibitors of protein–protein interactions tend to have certain properties that distinguish them from drugs that act against more conventional target classes. For example, they are larger and more three-dimensional.5,6 Therefore, it is an open and vital question whether fragments meant to serve as potential leads for protein–protein interaction targets should also have properties distinct from those of conventional fragments.
For selected PPI targets, the results of fragment screens have been reported,7 and hits have been described, but no overriding analysis has appeared comparing the properties of these PPI fragment hits to fragment hits from non-PPI systems. An answer to the question of what constitutes an optimal PPI fragment library will emerge as future drug discovery projects on this target class are pursued and reported upon. In the meantime, a complementary way of adding to our knowledge base is to perform retrospective analyses of successful programs. That is, to deconstruct known protein–protein inhibitors into successively smaller fragments and survey their potency and binding locations, and then compare these attributes to those of the parent compounds.
This strategy has already been applied.8−10 At Abbott, a very potent inhibitor of the Bcl-2 protein family was developed, designated ABT-737, and it ultimately entered the clinic as a potential cancer therapeutic. As commonly found for protein–protein inhibitors, its molecular weight, 813 Da, was substantially higher than what is commonly expected for a drug. In a retrospective study, compounds comprising portions of ABT-737 were obtained and were checked for activity, and the smallest piece that still exhibited binding was identified.8 The molecular weight of this smallest active fragment was 293 Da.
Interestingly, a plot of binding affinity vs molecular weight for this series of fragments produced a linear slope, and this relationship was confirmed in studies with additional targets. Therefore, one can use these data to predict the kinds of fragments that should be screened to find a good lead for a protein–protein interaction target. It was found that the affinities of the smallest active fragments were all in the range of 50–300 μM. If it is assumed that an acceptably potent drug candidate (1–10 nM) for a protein–protein target will have a typically high molecular weight (700–800 Da), then the fragment lead will need to have a molecular weight of about 300 Da, which is at the upper limit of the size range typical of fragment libraries.
In a related study, the Krimm group at the University of Lyon performed deconstruction analyses of ABT-737 and a variety of other published Bcl-2 family inhibitor scaffolds.9 While the previous Abbott study considered only scaffolds that were eventual successes, that is, were optimized into true drug candidates with desirable potency and PK properties, the Krimm study did not apply this restriction. In studies using ligand- and protein-based nuclear magnetic resonance (NMR) methods, binding was observed for several fragments derived from nine parent scaffolds, and some of these fragments possessed low molecular weights, in the range 120–230 Da.
A recent report10 described fragmentation of a small molecule inhibitor of the interaction between the von Hippel–Lindau protein (pVHL) and the alpha subunit of hypoxia-inducible factor 1 (HIF-1α), where the parent compound had a molecular weight of 410 Da. The study found that the smallest fragment that exhibited detectable binding by ligand-based NMR and thermal shift methods had a molecular weight of 262 Da. Further, binding could only be detected for fragments capable of occupying two adjacent subpockets at the interface. This collection of deconstruction studies of protein–protein systems is quite limited, so it is hard to draw practical generic conclusions, and consequently, guidelines for PPI fragment screening are currently undefined. On one hand, it appears that small (<200 Da) fragments can be found that bind to PPI targets. However, how promising are these fragment hits, that is, are they capable of evolving into bona fide drug candidates?
In order to contribute an example toward the understanding of how fragment size and structure relate to ultimate success in a drug discovery program, we have performed a deconstruction study of the Nutlins. The Nutlins constitute a distinct class of protein–protein inhibitor with a unique chemotype, and they have achieved high potency and successfully entered clinical trials.11 These compounds bind to the protein MDM2 and block its interaction with the multifunctional transcription factor p53. This enhances the overall level of p53 activity and thereby prevents cancer cells from evading apoptosis. In the study we report here, we have systematically deconstructed RG7112, the first member of the Nutlin family to enter clinical trials,12 into successively smaller fragments, and investigated the ability of these fragments to bind to MDM2 using surface plasmon resonance (SPR) (Figure 1), NMR (Figure 2), and X-ray crystallography (Figure 3). This investigation represents a valuable additional case toward answering the question: for a fragment library targeting protein–protein interactions, what key properties are shared by successful fragment leads?
Figure 1.

SPR sensograms (frame 1 on left) and plot of response vs concentration (frame 2 on right) for the binding of 1 to MDM2.
Figure 2.
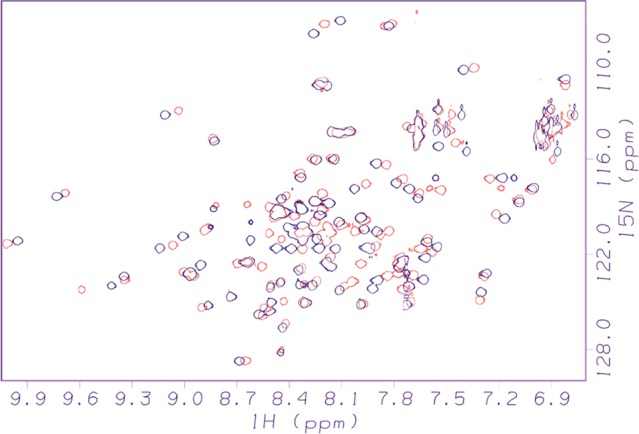
Representative 2D 1H–15N-HSQC NMR spectra, showing MDM2 in the empty state (black) and following the binding of 10 (red).
Figure 3.

X-ray crystal structures of MDM2 complexed with the parent Nutlin (1) (A) and various fragments: 9 (B); 10 (C); and 5 (D). In panel A, there is also a superimposed complexed p53 peptide14 depicted as a green ribbon with selected side chains shown as sticks.
The parent compound chosen for this study, RG7112 (1), is one of the most potent Nutlins developed to date, and its structure is shown in Table 1. Nutlins exert their inhibitory activity by binding to MDM2 and directly competing with the binding of the p53 protein. The binding of p53 to MDM2 can be fully replicated by a peptide fragment of p53 composed of residues 15–29.13 The X-ray structure of this peptide bound to MDM2 has been reported.14 It shows that, upon binding, the peptide adopts an alpha helical conformation and achieves affinity by inserting three hydrophobic side-chains from one side of this helix, Phe19, Trp23, and Leu26, into three corresponding subpockets of MDM2 (Figure 3A). An X-ray structure of 1 bound to MDM2 has been solved12 and establishes that 1, like all of the Nutlins, utilizes its tripod-like shape to efficiently insert substituents into these three subpockets. In the case of 1, the Phe, Trp, and Leu side-chains of p53 are effectively mimicked by, respectively, an ethoxy group and two chlorophenyl groups (Figure 3A).
Table 1. Structures, Molecular Weights, and MDM2 Binding Activity for RG7112 and Its Fragments.
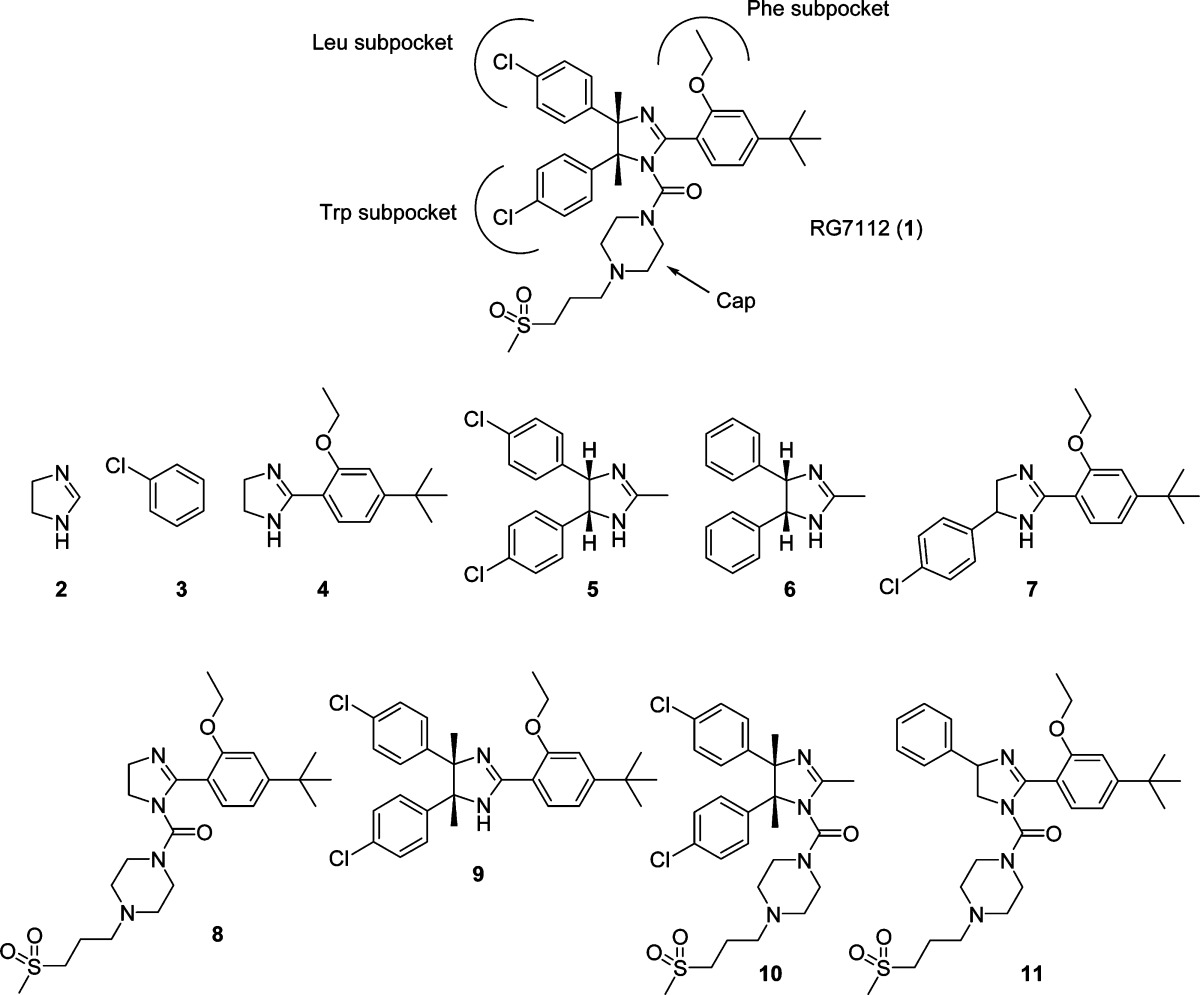
| compd | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MW | 727.78 | 70.09 | 112.56 | 246.35 | 305.2 | 236.31 | 356.89 | 478.65 | 495.48 | 565.56 | 554.75 |
| NMR binding? | yes | no | no | no | yes | no | yes | no | yes | yes | yes |
| SPR binding? (Kd, μM) | 0.22 | no | no | no | 26 | no | no | no | 20 | 14 | 1000 |
| ligand efficiency | 0.18 | 0.31 | 0.19 | 0.18 | 0.10 |
Nutlins also feature an appendage, in this case a piperazine derivative, extending from the N1 atom of the imidazoline core, which has been shown to exert a major influence on activity.17 This appendage typically does not make significant contact with the surface of MDM2, but rather projects outwardly into solvent. It is not obvious exactly how this moiety contributes to binding affinity, but its role may be to sterically direct a substituent into the Phe subpocket and/or to provide a cap that shields the hydrophobic interactions at the binding interface from solvent.
We have performed a systematic deconstruction of 1, obtaining compounds that represent successively smaller fragments of the parent (their structures are shown in Table 1). Because of practical limitations of chemical synthesis, not every conceivable fragment was achievable. However, this is a minor compromise, and the set of compounds provides an adequate coverage of the fragmentation pathways possible for 1.
For the set of fragments, SPR and NMR were applied independently to examine binding to MDM2. SPR was used to assess binding and to determine Kd values (Table 1) and off-rates. A representative SPR sensogram, showing binding of the parent compound to MDM2, is shown in Figure 1. In the SPR studies, the binding profiles for the noninteracting compounds were in general very distinct from the compounds that showed responses with MDM2, and nonspecific binding of the fragments to MDM2 was not observed. Two-dimensional 1H–15N-HSQC NMR was also used to assess and verify binding (Table 1). This form of protein-observe NMR is well established as one of the gold standard methods for sensitively detecting binding and for determining whether that binding is authentic, that is, does not involve unfolding, aggregation, or other undesirable effects on the protein. The HSQC method does not suffer from the higher false positive rates seen with SPR, ligand-observe NMR (in cases where verification by competition is not possible), and other biophysical techniques used to monitor binding in PPI systems. Furthermore, the pattern and magnitude of chemical shift changes observed in the HSQC experiment allows an experienced practitioner to judge between a binding mode based on significant insertion into the protein versus a more superficial type of binding involving only surface interactions.18 This latter form of binding, while resulting in classification as a hit in various methods, is unlikely to be capable of advancement toward improved potency. In addition, the HSQC NMR experiment provides a low-resolution footprint that indicates where on the target protein the ligand is interacting. This is important for determining if a fragment is binding in the same region it occupies when it is part of the intact parent compound. An exemplary superposition of two HSQC spectra, showing a spectrum of empty MDM2 overlaid with a spectrum acquired following the addition of one of the fragments, is pictured in Figure 2.
We also attempted X-ray crystallography for the fragments that were found to bind. While an NMR footprint is adequate for distinguishing whether a compound is located in the primary binding cleft or elsewhere, it is hard to determine the exact position of a compound and its orientation using chemical shift changes alone. X-ray crystallography can provide a precise picture of where and how the compound is bound to the protein. We were able to obtain structures for several of the fragments in complex with MDM2 via cocrystallization (Figure 3).
The Nutlin molecule can be conceptualized as an imidazoline core with four R groups, as depicted on the far right in Figure 4. There is a substituent (designated RTrp, RLeu, and RPhe) to occupy each of the three key subpockets of MDM2 (as shown schematically in Figure 1) and one substituent (RCap) that projects toward solvent and ostensibly caps the binding site. Our results show that when the parent Nutlin (1) was broken down to the level of individual R groups (compounds 3 and 4) or to the isolated imidazoline core (compound 2) none of these exhibited significant binding to MDM2, within the concentration limits of the SPR and NMR experiments (specific values are found in the Methods section of the Supporting Information). It appears that these fragments are too small to form sufficient interactions with the protein to sum up to a measurable binding affinity.
Figure 4.
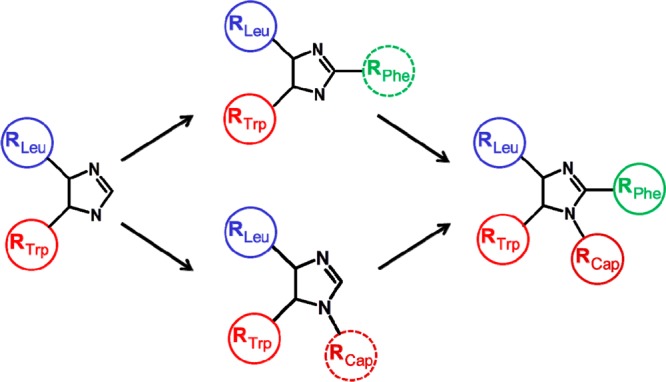
Hypothetical pathways by which a Nutlin could have been developed from a fragment hit, in accordance with the experimental data from the present study.
Toward the other extreme, fragments that retained three of the four R groups were all found to bind (compounds 9, 10, and 11). NMR indicated that these compounds were all binding in the active site cleft of MDM2. However, affinities varied among these fragments. Compound 10 comprising the Trp and Leu mimics coupled with the hydrophilic cap was the most potent, exhibiting a Kd of 14 μM. Similarly, the other fragment containing the Trp and Leu mimics, in this case coupled with the Phe mimic (9), was also relatively potent, exhibiting a Kd of 20 μM. The fragment containing the Phe mimic and the cap (11), with the Trp mimic added, in this case lacking the para-chloro substituent, was a much weaker binder, with a Kd of 1 mM. Nevertheless, this set of compounds indicates that a Nutlin fragment containing three R groups, in any combination, is capable of binding to MDM2. X-ray structures of compounds 9 and 10 bound to MDM2 (Figure 3B,C) show that these fragments attain a position as expected based on the Nutlin binding paradigm, exhibiting the same orientation and utilizing the same subpocket-filling strategy as the parent (Figure 3A).
The binding of fragments that retained only two of the four R groups was more varied. A fragment consisting of the combination of the Phe mimic and the cap (8) did not bind significantly to MDM2. However, a fragment combining the Trp mimic and the Leu mimic (5) was able to bind. NMR indicated that the binding of this fragment was authentic and that it was located in the active site cleft. The affinity of this compound, which was found to be 26 μM, was not far from that exhibited by fragments possessing three R groups. X-ray crystallography verified that 5 maintains the binding strategy of the parent, as it inserts the expected substituents into the Trp and Leu subpockets (Figure 3D). A fragment (7) was produced that combined one chlorophenyl substituent with the Phe mimic; because of rapid interconversion, the chlorophenyl would be capable of occupying either the Trp or Leu subpocket. This compound produced a chemical shift perturbation in NMR indicative of binding to MDM2, with its location in the active site cleft. However, the binding was apparently too weak to be confirmed by SPR performed at the testable limit of 1 mM. Correspondingly, attempts to obtain cocrystals of this compound with MDM2 were unsuccessful. Overall, this set of compounds established that Nutlin fragments containing two of the four parent R groups were capable of binding to MDM2. The particular combination of R groups was found to have a dramatic influence on activity, causing affinities to range from an undetectable level to a Kd of 26 μM.
The ligand efficiency (LE)15 value of 0.31 found for fragment 5 is within the range recommended for an acceptable fragment hit.16 However, expansion to the larger fragments 9, 10, and 11 resulted in a drop to LE values of 0.10–0.19, and although a commonly accepted goal during drug optimization is to maintain LE values near 0.3, these values are comparable to those of the parent Nutlin, which despite its low efficiency possessed all of the properties needed for entry into clinical trials.
The conformation of the MDM2 protein was basically the same among all of the complexed structures, with the exception of the area near the Phe subpocket. As can be seen in Figure 3, when the bound ligand has an appendage filling the Phe subpocket, Tyr63 of MDM2 is flipped away, and Met58 is oriented inward (Figure 3A,B); while, when the ligand is lacking a Phe mimic, Tyr63 flips inward and Met58 is pushed away (Figure 3C,D).
Having identified 5 as the smallest Nutlin fragment capable of binding to MDM2, we investigated whether a trimmed-down version of 5 would still be able to bind. A derivative of 5 was prepared, which lacked the para-chloro substituents (compound 6). It was found to be incapable of binding to MDM2. This underscores the high importance of the para-chloro substituents in the dissection of binding determinants for RG7112.
The molecular weight of the smallest fragment of Nutlin that retains binding competency was established at 305 Da. This value is at the high end of the molecular weight range that normally defines fragments. This finding compares closely with that obtained for ABT-737, another small molecule inhibitor of a protein–protein interaction that has achieved clinical entry. Further, the finding that the smallest Nutlin fragment competent to bind occupies two adjacent subpockets on MDM2 is consistent with the earlier deconstruction study of the pVHL inhibitor,10 where only fragments capable of accessing two subpockets exhibited detectable binding. While this pair represents a small sample size, there may be a trend emerging with respect to the composition of fragment screening libraries aimed against protein–protein interaction targets, namely, it may be advantageous to skew them toward higher molecular weights. As fragment screening is performed increasingly against protein–protein systems, it will be educational to collectively analyze the results to see if this trend is subtantantiated. Since the purpose of employing small compounds is to more efficiently sample chemical space, an increase in size is undesirable as it will offset this advantage by requiring many more compounds to achieve suitable coverage. Nevertheless, this may turn out to be a necessary trade-off.
In retrospect, it appears that the Nutlin series of MDM2 inhibitors could have been discovered via a fragment-based approach (Figure 4), although the library would require compounds with molecular weights over 300 Da. One hypothetical pathway would start with detection of binding of compound 5 by NMR, SPR, or some other method, and verification as a true binder by HSQC NMR. Subsequent chemical elaboration could have led to one of the versions containing three R groups, all of which were found to be capable of binding. The addition of the fourth R group would have resulted in a complete and highly potent Nutlin. The optimization process could have been guided throughout by X-ray structures, as the initial hit and all the key derivatives along the pathway were found to be competent for cocrystallization with MDM2.
In conclusion, our study supports the notion that protein–protein interaction systems should be highly amenable to a fragment-based lead discovery approach, although these systems will likely require some specialized choice of library composition.
Acknowledgments
We gratefully acknowledge the efforts of Santina Russo and Joachim Diez of Expose GmbH in collecting the diffraction data at the SLS.
Supporting Information Available
Experimental methods for chemical synthesis, SPR, NMR, and X-ray crystallography. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): The authors are all current or former employees of Hoffmann-La Roche, Inc.
Supplementary Material
References
- Wells J. A.; McClendon C. L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 2007, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Fry D. C.Targeting Protein–Protein Interactions for Drug Discovery. In Protein–Protein Interactions: Methods and Applications, 2nd ed.; Fu H., Meyerkord C., Eds.; Springer: New York, 2013; in press. [Google Scholar]
- Murray C. W.; Rees D. C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51, 4990–5003. [DOI] [PubMed] [Google Scholar]
- Sperandio O.; Reynes C. H.; Camproux A. C.; Villoutreix B. O. Rationalizing the chemical space of protein–protein interaction inhibitors. Drug Discovery Today 2010, 15, 220–229. [DOI] [PubMed] [Google Scholar]
- Fry D. C.; So S.-S.. Modulators of Protein–Protein Interactions: The Importance of Three-Dimensionality. In Protein–Protein Interactions in Drug Discovery; Doemling A., Ed.; Wiley-VCH: Weinhelm, Germany, 2012; pp 55–62. [Google Scholar]
- Bower J. F.; Pannifer A. Using fragment-based technologies to target protein–protein interactions. Curr. Pharm. Des. 2012, 18, 4685–4696. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J. Fragment-based drug design: how big is too big?. J. Med. Chem. 2006, 49, 6972–6976. [DOI] [PubMed] [Google Scholar]
- Barelier S.; Pons J.; Marcillat O.; Lancelin J.-M.; Krimm I. Fragment-based deconstruction of Bcl-XL inhibitors. J. Med. Chem. 2010, 53, 2577–2588. [DOI] [PubMed] [Google Scholar]
- Van Molle I.; Thomann A.; Buckley D. L.; So E. C.; Lang S.; Crews C. M. Dissecting fragment-based lead discovery at the von Hippel–Lindau Protein:Hypoxia Inducible Factor 1α protein–protein interface. Chem. Biol. 2012, 19, 1300–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev L. T.; Vu B. T.; Graves B.; Carvajal D.; Podlaski F.; Filipovic Z.; Kong N.; Kammlott U.; Lukacs C.; Klein C.; Fotouhi N.; Liu E. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Tovar C.; Graves B.; Packman K.; Higgins B.; Xia M.; Tardell C.; Garrido R.; Lee E.; Kolinsky K.; To K.-H.; Linn M.; Podlaski F.; Wovkulich P.; Vu B.; Vassilev L. MDM2 small molecule agonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [DOI] [PubMed] [Google Scholar]
- Picksley S. M.; Vojtesek B.; Sparks A.; Lane D. P. Immunochemical analysis of the interaction of p53 with MDM2: fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene 1994, 9, 2523–2529. [PubMed] [Google Scholar]
- Kussie P. H.; Gorina S.; Marechal V.; Elenbaas B.; Moreau J.; Levine A. J.; Pavletich N. P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Schultes S.; de Graaf C.; Haaksma E. E. J.; de Esch I. J. P.; Leurs R.; Kramer O. Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discovery Today Technol. 2010, 7, e157–e162. [DOI] [PubMed] [Google Scholar]
- Vu B.; Wovkulich P.; Pizzolato G.; Lovey A.; Ding Q.; Jiang N.; Liu J.-J.; Zhao C.; Glenn K.; Wen Y.; Tovar C.; Packman K.; Vassilev L.; Graves B. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013, 4, 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson M. P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. 2013, 73, 1–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.