

Abstract
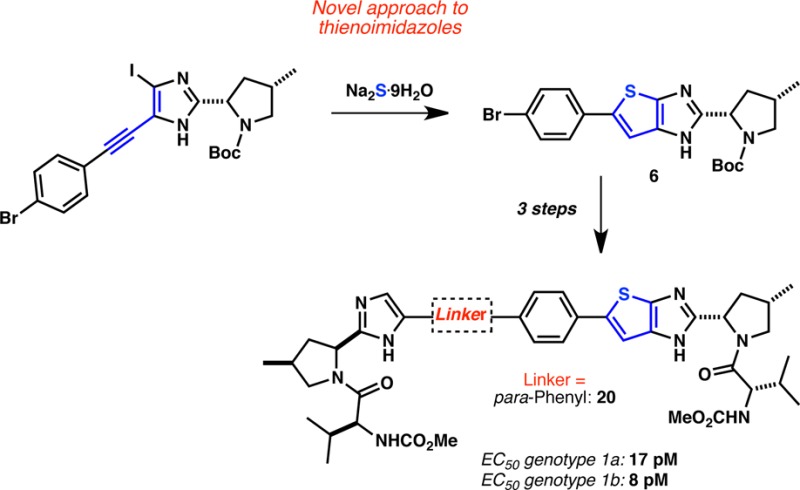
The discovery of potent thienoimidazole-based HCV NS5A inhibitors is herein reported. A novel method to access the thienoimidazole [5,5]-bicyclic system is disclosed. This method gave access to a common key intermediate (6) that was engaged in Suzuki or Sonogashira reactions with coupling partners bearing different linkers. A detailed study of the structure–activity relationship (SAR) of the linkers revealed that aromatic linkers with linear topologies are required to achieve high potency for both 1a and 1b HCV genotypes. Compound 20, with a para-phenyl linker, was identified as a potential lead displaying potencies of 17 and 8 pM against genotype 1a and 1b replicons, respectively.
Keywords: HCV, NS5A, thienoimidazoles
The hepatitis C virus (HCV) is a pathogen currently affecting 170 million people worldwide.1 Since the discovery of HCV in 1989, intense research efforts to find a cure have been pursued on both the academic and the pharmaceutical fronts.2,2b The standard of care (SOC) has recently evolved from a two-drug combination of intravenous pegylated interferon-α (PEG-IFN) and oral ribavirin (RBV)3 to a three-drug combination of PEG-IFN, RBV, plus one of two recently approved direct-acting antivirals (DAAs), Incivek or Victrelis. These new DAAs act as reversible covalent inhibitors of the NS3/4A protease.4−4d When combined with PEG-IFN/RBV, either of these two agents can significantly increase sustained virological response (SVR) and dramatically reduce treatment duration.5 However, new treatments with all-oral regimens that would eliminate the need for intravenous PEG-IFN are desirable.
To this end, the nonstructural protein NS5A has recently emerged as a potential therapeutic target for HCV.6−7b Although NS5A has no known enzymatic activity and its exact role remains elusive, evidence suggests that it plays an essential function in the regulation of the HCV replication.6
To date, several NS5A inhibitors are in various stages of development, most notably, daclatasvir (BMS-790052, 1, Figure 1), which has emerged as the benchmark molecule in this area.7,7b In clinical trials, 1 displayed an impressive 3 log10 reduction of HCV RNA in 24 h with a 10 mg dose (genotypes 1a and 1b).8
Figure 1.
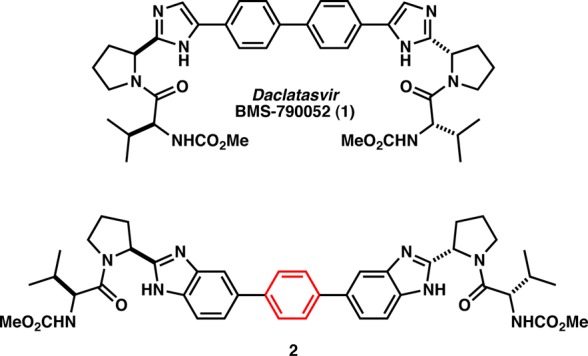
Chemical structures of BMS-790052 (1) and 2.
C2 symmetric bis-benzimidazole analogues with aryl linkers, such as 2, have been shown to be potent inhibitors of NS5A in both genotype 1a and 1b replicons.9 In this manuscript, we describe our results at replacing one of the benzimidazoles with an isosteric thienoimidazole moiety, thereby breaking the C2 symmetry of the molecules. We also provide the details of the linker structure–activity relationship (SAR) studies of two unsymmetrical series, namely, the benzimidazole and the imidazole series.
Synthetically, we envisaged that a disconnection involving the coupling of a linker bearing an aryl boronic acid (Suzuki) or an acetylene derivative (Sonogashira) with a common intermediate (6) could provide us a versatile method giving access to the full carbon framework of these analogues. Finally, a two-step sequence involving an acid-induced N-Boc deprotection and the capping of both pyrrolidine nitrogens with the requisite valine carbamate moieties would give access to the desired analogues. Our synthetic approach is presented in Figure 2.
Figure 2.
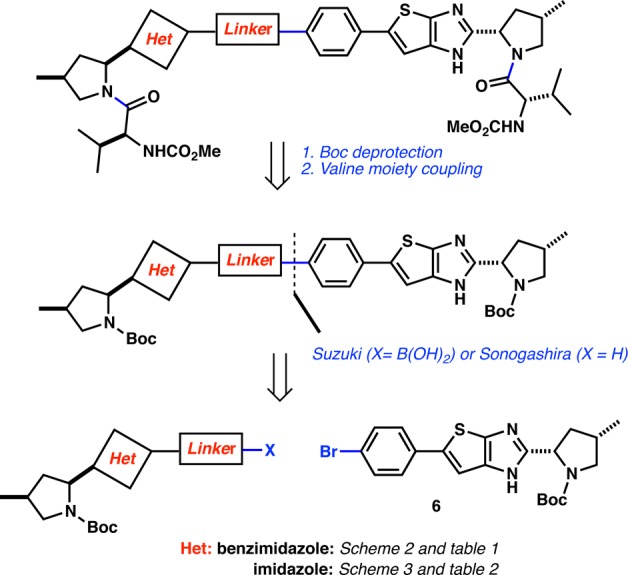
Retrosynthetic analysis.
First, we undertook the synthesis of the thienoimidazole key intermediate 6 following the sequence in Scheme 1. To this end, imidazole 3(10) was converted to iodoimidazole 4 via a bis-iodination using N-iodosuccinimide and a proto-dehalogenation in 72% yield over two steps. Iodoimidazole 4 was coupled to 4-bromophenylacetylene under the Sonogashira conditions, and the resulting intermediate was smoothly iodinated with N-iodosuccinimide to afford alkyne 5 in 76% yield over two steps. The key cyclization leading to the required thienoimidazole core was then attempted. Much to our delight, thienoimidazole 6 could be isolated in ca. 40% yield after exposure of 5 to Na2S·9H2O and CuI in DMSO. To the best of our knowledge, this reaction represents a novel entry to the strained [5,5]-bicyclic thienoimidazole core.11
Scheme 1. Preparation of Key Intermediate 6 via a Novel Entry to the Thienoimidazole Core.

Reagents and conditions: (a) NIS, CH2Cl2, 0 °C to RT. (b) LiCl, MeMgCl, i-PrMgCl, THF, −20 °C to RT (c) 4-Bromophenylacetylene, CuI, PdCl2·(dppf), NEt3, DMF, 80 °C. (d) NIS, CH2Cl2, RT. (e) Na2S·9H2O, CuI, DMSO, 150 °C.
The key intermediate 6 was then used to synthesize all compounds described herein. Generally, a three-step sequence gave access to all final compounds:12 (1) Suzuki or Sonogashira coupling of the corresponding partner, (2) N-Boc deprotection, and (3) coupling of the required valine N-methylcarbamate moiety7,7b,9 (Schemes 2 and 3).
Scheme 2. General Preparations of NS5A Inhibitors 7–10 via the Suzuki and Sonogashira Reactions.
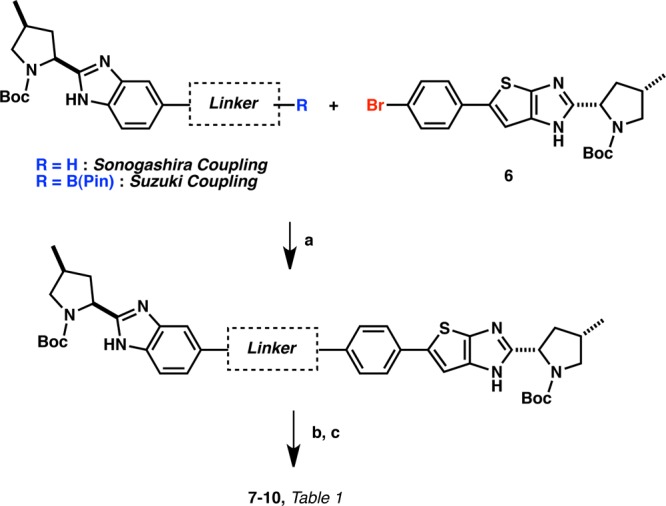
Reagents and conditions: (a) Pd(OAc)2, sodium 2′-dicyclohexylphosphino-2,6-dimethoxy-1,1′-biphenyl-3-sulfonate hydrate, NaHCO3, IPA/H2O, 120 °C, μW. (b) TFA, CH2Cl2 or HCl, dioxane, RT. (c) t3p, DIEA, (S)-N-methoxycarbonyl valine, CH2Cl2.
Scheme 3. General Preparations of NS5A Inhibitors 11–21 via the Suzuki Reaction.
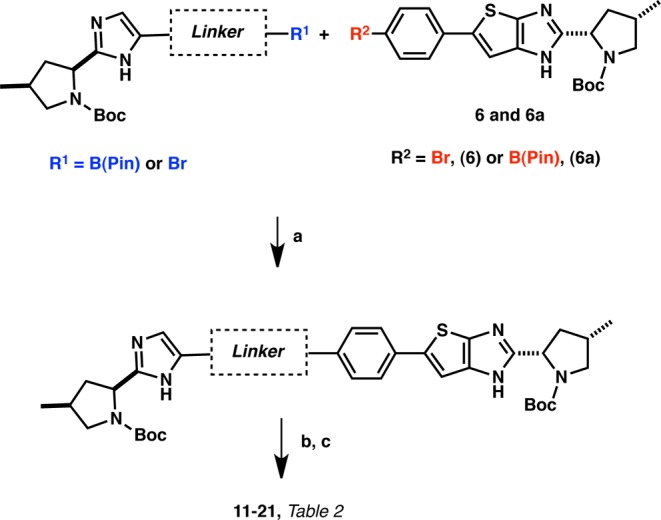
Reagents and conditions: (a) Pd(OAc)2, sodium 2′-dicyclohexylphosphino-2,6-dimethoxy-1,1′-biphenyl-3-sulfonate hydrate, NaHCO3, IPA/H2O, 120 °C, μW. (b) TFA, CH2Cl2 or HCl, dioxane, RT. (c) t3p, DIEA, (S)-N-methoxycarbonyl valine, CH2Cl2.
Compounds 7–10 are unsymmetrical benzimidazole-thienoimidazoles (Table 1). Excellent genotype 1b (G1b) replicon potency was achieved with or without a linker, and good to modest genotype 1a (G1a) replicon potency was observed for compounds 7–9 (Table 1). However, the addition of the ethylene linker resulted in a 16-fold enhancement of G1a potency (0.038 nM, 10) as compared to 7 that bears no linker (0.619 nM, 7). We postulate that the ethylene linker of 10 could facilitate the optimal orientation of the essential valine-proline moieties for optimal contact with the G1a protein. The flexibility of 10 as compared to the more rigid and linear arrangement of phenyl (8) and acetylene (9) derivatives may be responsible for the increased G1a activity.
Table 1. SAR Studies of the Linker Moiety (Benzimidazole Series).
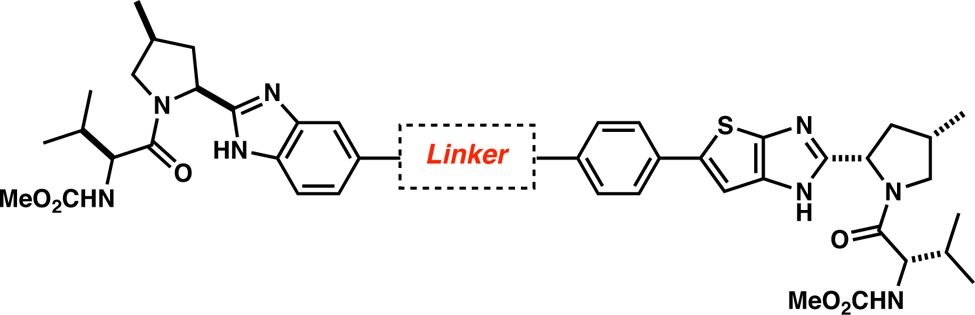
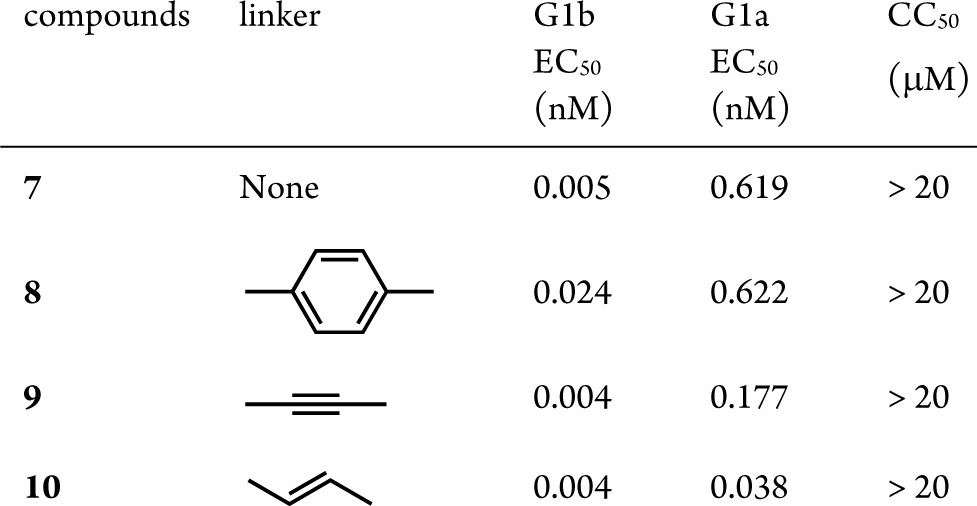
To assess if additional flexibility around the benzimidazole moiety is a requirement to achieve G1a potency, we replaced the [5,6]-benzimidazole system with an imidazole, directly attached to different linkers (Scheme 3). The change of the benzimidazole moiety for an imidazole would give rise to compounds with topologies reminiscent of BMS-790052 (1, G1a = 50 pM, G1b = 9 pM) and ultimately could lead to better activities against genotype 1a.
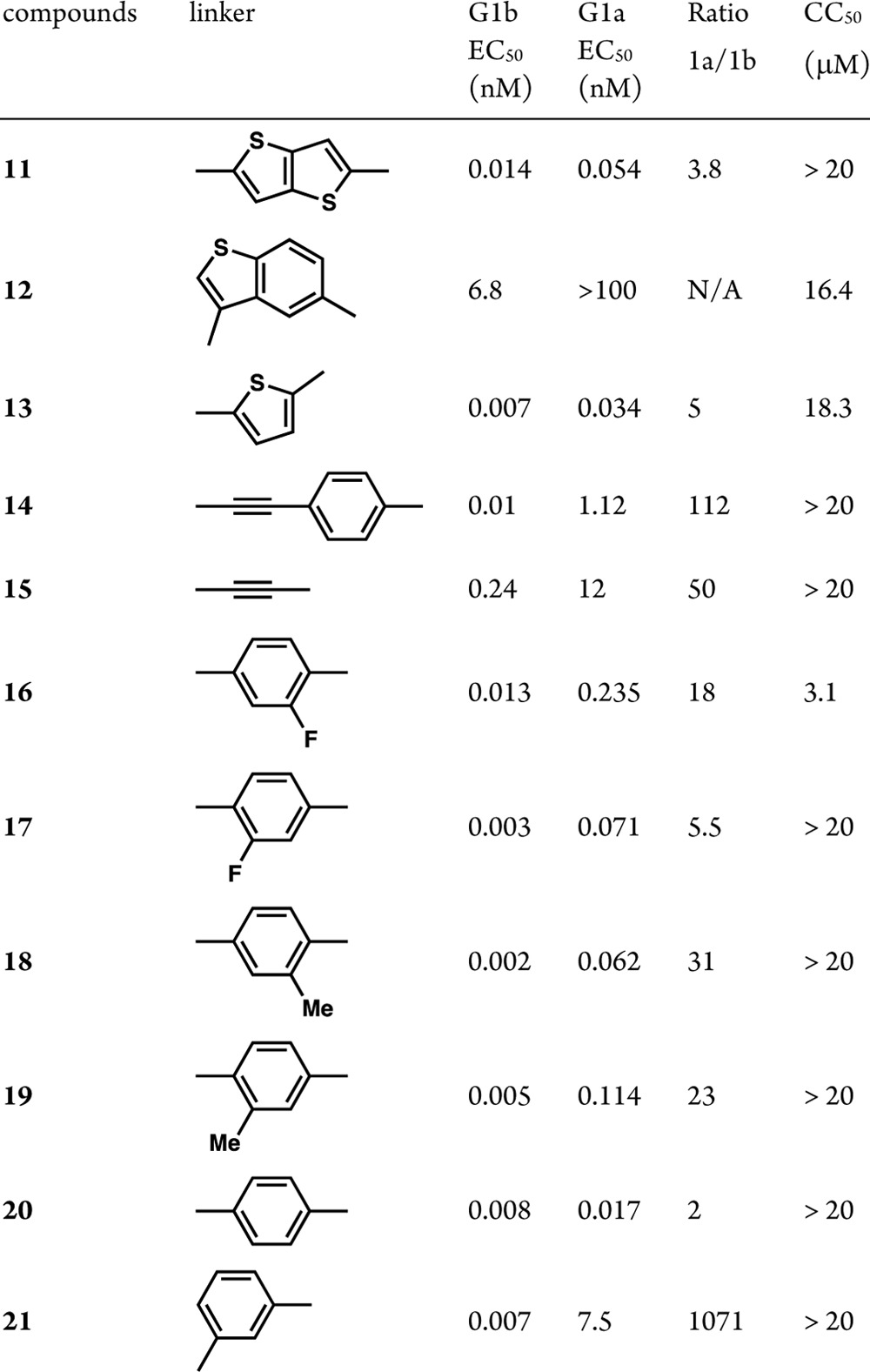
Indeed, very strong G1a potencies (EC50 < 0.1 nM) could be achieved with a bis-fused thiophene, thiophene, and phenyl-based linker (Table 2). Remarkably low ratios of 1a/1b (≤6) were obtained for 11, 13, 17, and 20 with compound 20 displaying replicon EC50 values of 0.008 and 0.017 nM against G1b and G1a, respectively. The para arrangement of the linker seems to be critical as the meta-substituted phenyl linker displayed very low potency against G1a (compare compounds 20 and 21), further confirming the critical spatial arrangement of the proline-valine moiety required to achieve G1a potency. We postulate that these compounds bind at the dimeric NS5A interface, thereby preventing the formation of the active NS5A homodimer. This is supported by the identification of mutations in the replicon assay (G1a: M28, Q30, L31, and Y93; G1b: Y93), which are proximal to or at the dimer interface.13 These mutations have also been reported for BMS-790052 (1) and other NS5A inhibitors.6,14 Additionally, all analogues from Tables 1 and 2 were noncytotoxic displaying CC50 values greater than 20 μM.15
Table 2. SAR Studies of the Linker Moiety (Imidazole Series).
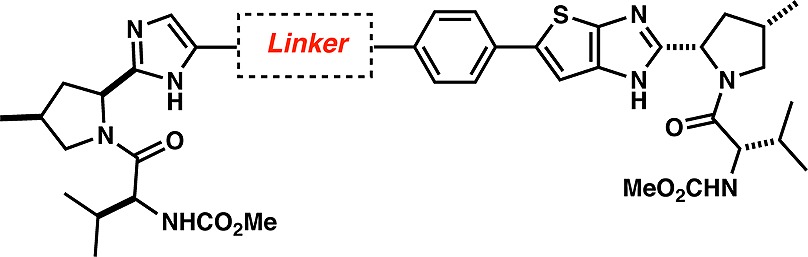
With good G1a and G1b potencies in hand, the ADME properties of 20 were investigated further. Metabolic stability was evaluated in rat (RLM) and human (HLM) liver microsomes in the presence of NADPH and UDPGA as cofactors. Compound 20 was stable in rat and human microsomes with 91 and 100% parent intact, respectively, after incubation for 1 h at a concentration of 5 μM. The observed Caco-2 ratio (BA/AB ratio) of 2.0 suggested that compound 20 is not a substrate for the Pgp efflux transporter. Importantly, the pharmacokinetic profile of 20 in rats indicated low clearance (Cl 5.3 mL/min/kg) and moderate bioavailability (F = 25%) after dosing (3 mg/kg) as a solution in 30% aqueous PEG-400. Key pharmacokinetic data for selected compounds are included in Table 3. Compounds 10, 11, and 21 also display low Cl below 15 mL/min/kg and half-lives (T1/2) ranging from 5 to 11 h.
Table 3. Pharmacokinetic Profiles for Selected Compounds.
| compds |
||||
|---|---|---|---|---|
| 10 | 11 | 20 | 21 | |
| iv rat PK | ||||
| Cl (mL/min/kg) | 6.0 | 5.1 | 5.3 | 13.5 |
| T1/2 (h) | 6.6 | 5.1 | 6.1 | 5.1 |
| Vss (L/kg) | 2.7 | 2.0 | 2.4 | 4.8 |
| po rat PK | ||||
| AUC0–8h (μg h/mL) | 0.82 | 0.46 | 1.54 | 0.49 |
| T1/2 (h) | 5.0 | 10.8 | 8.3 | 5.1 |
| Cmax (ng/mL) | 0.03 | 0.01 | 0.03 | 0.01 |
| % F | 14.1 | 6.7 | 25.6 | 16 |
In summary, we describe the identification and optimization of two series of nonsymmetrical thienoimidazole-containing analogues with subnanomolar potency against both genotypes 1a and 1b replicons. This study demonstrated that benzimidazoles can successfully be replaced by the novel thienoimidazole isostere when a proper linker is chosen. The optimization of compound 20, geared toward improved potency against the mutations identified in the replicon assay, will be reported in due course.
Acknowledgments
We thank James Empfield and Rene Rijnbrand for useful comments on the manuscript as well as Ding Lu and Gregory May for assistance on characterization.
Supporting Information Available
Full experimental details for representative compounds synthesized and description of assays. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Negro F.; Alberti A. The global health burden of hepatitis C virus infection. Liver Int. 2011, 31, 1–3. [DOI] [PubMed] [Google Scholar]
- For reviews, see; De Francesco R.; Migliaccio G. Challenges and successes in developing new therapies for hepatitis C. Nature 2005, 436, 953. [DOI] [PubMed] [Google Scholar]
- Houghton M.; Abrignani S. Prospect for a vaccine against the hepatitis C virus. Nature 2005, 436, 961–966. [DOI] [PubMed] [Google Scholar]
- Feld J. J.; Hoofnagle J. H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [DOI] [PubMed] [Google Scholar]
- Pearlman B. L. Protease inhibitors for the treatment of chronic hepatitis C genotype-1 infection: the new standard of care. Lancet Inf. Dis. 2012, 12, 717–728. [DOI] [PubMed] [Google Scholar]
- Chatel-Chaix L.; Baril M.; Lamarre D. Hepatitis C Virus NS3/4A Proteases Inhibitors: A Light at the End of the Tunnel. Viruses 2010, 2, 1752–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. X.; Njoroge F. G. A review of HCV protease inhibitors. Curr. Opin. Invest. Drugs 2009, 10, 821–837. [PubMed] [Google Scholar]
- Kwong A. D.; McNair L.; Jacobson I.; George S. Recent progress in the development of selected hepatitis C virus NS3/4A protease and NS5B polymerase inhibitors. Curr. Opin. Pharmacol. 2008, 8, 522–531. [DOI] [PubMed] [Google Scholar]
- Poordad F.; Dieterich D. Treating hepatitis C: Current standard of care and emerging direct-acting antiviral agents. J. Viral Hepatitis 2012, 19, 449–464. [DOI] [PubMed] [Google Scholar]
- Schmitz U.; Tan S.-L. NS5A—From Obscurity to New Target for HCV Therapy. Recent Pat. Anti-infect. Drug Discovery 2008, 3, 77–92. [DOI] [PubMed] [Google Scholar]
- Lemm J. A.; O’Boyle D. II; Liu M.; Nower P. T.; Colonno R.; Deshpande M. S.; Snyder L. B.; Martin S. W.; St. Laurent D. R.; Serrano-Wu M. H.; Romine J. L.; Meanwell N. A.; Gao M. Identification of Hepatitis C Virus NS5A Inhibitors. J. Virol. 2010, 84, 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell T. W. Drugs for Hepatitis C: Unlocking a New Mechanism of Action. ChemMedChem 2010, 5, 1663–1665. [DOI] [PubMed] [Google Scholar]
- Gao M.; Nettles R. E.; Belema M.; Snyder L. B.; Nguyen V. N.; Fridell R. A.; Serrano-Wu M. H.; Langley D. R.; Sun J.-H; O’Boyle D. R. II; Lemm J. A.; Wang C.; Knipe J. O.; Chien C.; Colonno R. J.; Grasela D. M.; Meanwell N. A.; Hamann L. G. Chemical genetics strategy identifies an HCV NS5A inhibitor with potent clinical effect. Nature 2010, 465, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira O.; Maxwell J.; Bennani Y. L.; Das S. K.; Kong L. C. C.; Reddy T. J.; Giroux S.; Cottrell K. M.; Morris M. A. Patent WO/2011/119858.
- For the synthesis, see Kong L. C. C.; Giroux S.; Reddy T. J; Bennani Y. L. Patent WO/2011/119870 and reference therein.
- For an alternative synthesis of the thienoimidazole core, seeHartley D. J.; Iddon B. Use of the Vinyl Group as an Efficient Protecting Group for Azole N-Atoms: Synthesis of Polyfunctionalized Imidazoles and Thieno[2,3-d]-[3,2-d]imidazole. Tetrahedron Lett. 1997, 38, 4647–4650. [Google Scholar]
- See the Supporting Information for the details of the syntheses of intermediate 6 and compound 20.
- Large potency drops were observed when 20 was screened against recombinant replicon cell lines harboring some of these mutations: (1a Y93H/C, >15000-fold; 1a M28T and Q30R, >500-fold; and 1b Y93H, 900-fold).
- Fridell R. A.; Qui D.; Wang C.; Valera L.; Gao M. Resistance Analysis of the Hepatitis C Virus NS5A Inhibitor BMS-790052 in an In Vitro Replicon System. Antimicrob. Agents Chemother. 2010, 54, 3641–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CC50 in ET cells were determined using a [3H]-thymidine incorporation assay from 12 concentrations of test compound in duplicate, and data were considered accurate when the CC50 of the positive control was within 40% of the mean value.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.