

Abstract
Activating mutations in the epidermal growth factor receptor (EGFR) have been identified in a subset of non-small cell lung cancer (NSCLC), which is one of the leading cancer types worldwide. Application of EGFR tyrosine kinase inhibitors leads to acquired resistance by secondary EGFR mutations or by amplification of the hepatocyte growth factor receptor (c-Met) gene. Although several EGFR and c-Met inhibitors have been reported, potent dual EGFR/c-Met inhibitors, which can overcome this latter resistance mechanism, have hitherto not been published and have not reached clinical trials. In the present study we have identified dual EGFR/c-Met inhibitors and designed novel N-[4-(quinolin-4-yloxy)-phenyl]-biarylsulfonamide derivatives, which inhibit the c-Met receptor and both the wild-type and the activating mutant EGFR kinases in nanomolar range. We have demonstrated by Western blot analysis that compound 10 inhibits EGFR and c-Met phosphorylation at cellular level and effectively inhibits viability of the NSCLC cell lines.
Keywords: NSCLC, EGFR, c-Met, kinase inhibitor, acquired resistance
Identification of oncogene-driven signaling pathways harnessed by tumor cells and their inhibition by small-molecules is a key process for drug development resulting in new medicines such as EGFR inhibitor gefitinib, erlotinib, or lapatinib.1 EGFR-related signaling pathways play an important role in several malignant processes including NSCLC and have been proven to be potential therapeutic targets.2 Examination of mutations in the EGFR domain is a useful tool for predicting efficacy of EGFR TKIs.3,4 Those tumor cells, which harbor activating mutations in the EGFR kinase domain (e.g., point mutation L858R or in-frame deletions), respond impressively to current EGFR TKIs;5 nevertheless, the emergence of secondary mutations in the EGFR gene eliminates the efficacy of these drugs.6 Nearly half of all acquired resistance is due to the secondary T790M point mutation in exon 207−9 on the EGFR gene.
Another important oncogene in NSCLC is the hepatocyte growth factor (HGF) receptor also known as scatter factor (SF) or c-Met and its natural ligand, HGF.10 The receptor tyrosine kinase c-Met is implicated in many cellular processes such as proliferation, survival, migration, invasion, and wound healing.11 Amplification of the c-Met gene causes secondary resistance against EGFR TKI therapy in 18–20% of the cases, and it can occur parallel with T790M EGFR mutation.12,13 Pharmacological inhibition of c-Met kinase restores sensitivity of both T790M and c-Met gene amplification-derived resistant lung cancer cells to EGFR TKIs.14 On the basis of these results, the simultaneous inhibition of EGFR and c-Met kinases seems desirable; however, to date only the quinazoline-based MJ-56 has been reported to decrease both c-Met and EGFR phosphorylation in HT-29 colorectal cancer cells at relatively high concentration (15 μmol).15 Potent EGFR/c-Met dual inhibitors have not been developed until now and no such drug candidates are in clinical trials, but several EGFR and c-Met selective inhibitors have been reported as drug candidates or are in clinical development.16,17 In the present study, we report novel small-molecule kinase inhibitors that inhibit both EGFR and c-Met activity at nanomolar range in enzymatic assays and induce apoptosis in a clinically relevant HCC827 NSCLC cell line. As the initial step, our in-house compound collection of Nested Chemical Library was screened by recombinant wt EGFR and c-Met kinase assays, and we have identified 1 and 2 as very effective c-Met inhibitors. These compounds were ineffective on EGFR while strongly inhibiting InsR, which is considered undesirable. Potent AXL inhibitory effect has been reported for (4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide scaffold of BMS-777607 (IC50: 1.1 nM) and quinoline-based sulphonamides (IC50: 16 nM).18 Taking the structural similarity between c-Met and AXL kinases into account, we assumed that the antipyrine-carboxamide motif is also bioisostere with the biaryl-sulfonamide scaffold in regard to c-Met (Figure 1). To verify this, we performed docking simulations that led to effective biaryl-sulfonamide derivatives against c-Met (Supporting Information). In vitro assays of the biaryl-sulfonamide analogues demonstrated increased EGFR inhibition and decreased c-Met and InsR inhibition compared to 1 and 2.
Figure 1.
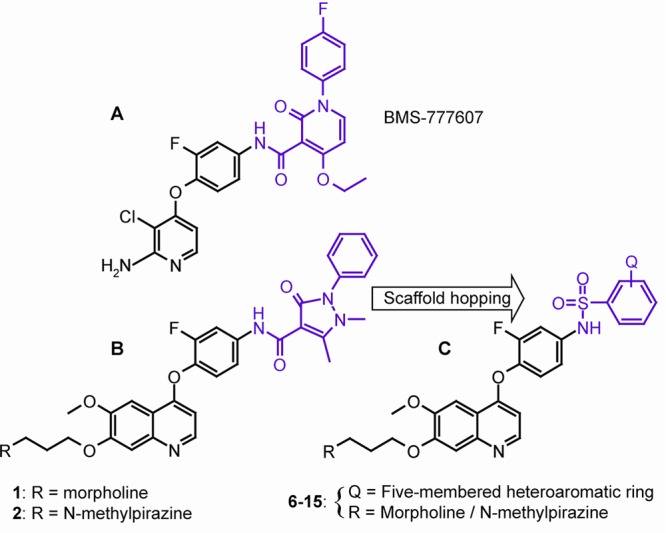
Structures of the identified inhibitor and its analogues. (A) Structure of c-Met inhibitor BMS-777607. (B) Structure of the original hits, compounds 1 and 2. (C) General structure of the EGFR/c-Met inhibitors, compounds 6–15.
To examine the structure–activity relationship (SAR), we prepared further derivatives (6–15) regarding to the previously reported synthetic routes of c-Met inhibitors (Table 1).19−21 In our synthetic strategy we applied a similar method22,23 to built up side-chain fitted quinoline derivatives affording the 3a–b nitro compounds. It was reduced by catalytic hydrogenation affording the amine intermediates 4a–b, which were reacted with aromatic sulfonyl-chlorides containing bromine providing 5a–c. These were cross-coupled with five-membered heteroaromatic boronic acids under palladium-catalyzed Suzuki-Myaura conditions24 using microwave irradiation at 140 °C in 1,2-dimethoxyethane to give biaryl derivatives in moderate yield (Scheme 1).
Table 1. Stucture–Activity Relationship, Enzymatic Data, and Cell Viability Inhibition of N-[4-(Quinolin-4-yloxy)-phenyl]-biarylsulfonamidesa.
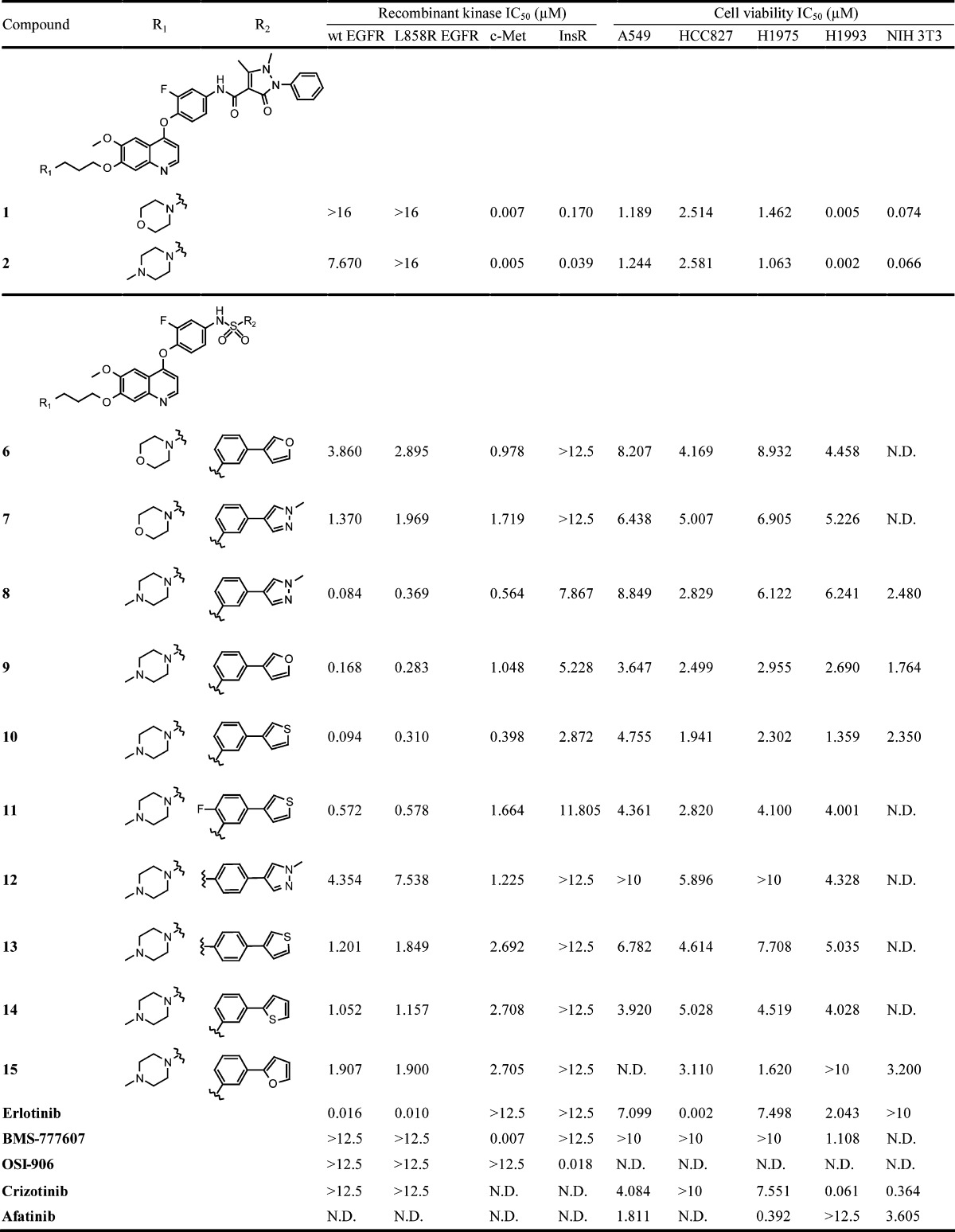
Enzyme activity and cell viability inhibition. IC50 values (μM) of the compounds on four recombinant enzymes, on various NSCLC, and on the NIH 3T3 cell lines. Cell viability data were gathered with MTT assay. All values are an average of at least three independent experments. N.D., not determined.
Scheme 1. Synthesis Route of N-[4-(Quinolin-4-yloxy)-phenyl]-biarylsulfonamides.

Reagents and conditions: (a) methanol/10% Pd/C, rt; (b) anhydrous pyridine, 3- or 4-Br-PhSO2Cl, 50 °C, 24 h; (c) Pd(0)PPh4, Na2CO3, Ar–B(OH)2, 140 °C, μW, (20–40%).
By comparing the analogues, we found 8 (wt EGFR: 84 nM, c-Met: 564 nM), 9 (wt EGFR, 168 nM; c-Met, 1048 nM), and 10 (wt EGFR, 94 nM; c-Met, 398 nM) derivatives to be the most effective against EGFR and c-Met kinases, while the corresponding 4-substitued analogues showed less potency both in enzymatic and cellular assays. The quality of the side chain is also pivotal for the efficacy of these compounds: especially, the 3-(4-methylpiperazin-1-yl)-propoxy side-chain enhanced the inhibitory effect dramatically (e.g., wt EGFR, 6 3860 nM vs 9 168 nM; wt EGFR, 7 1370 nM vs 8 84 nM), which prompted us to focus on it in our synthetic efforts. Three compounds were selected for further characterization as potential dual EGFR/c-Met inhibitors, and compound 10 was found the most effective and therefore selected for testing in various biochemical and cellular assays. To investigate the selectivity of this series, compound 10 was screened at 1 μM concentration on a recombinant kinase selectivity panel of 34 clinically relevant kinases, and it showed more than 70% inhibition against only six other receptor tyrosine kinases: DDR1 (111%), AXL (109%), c-KIT (91%), ErbB2 (81%), RET (78%), and FLT3 (76%) (see Supporting Information). On the basis of the preliminary data, it was hypothesized that all the compounds were ATP-analogue, reversible kinase inhibitors, and to assess whether the molecules bind the ATP-binding cleft in the kinases, we analyzed the enzyme activity data on double reciprocal plots. The trendlines intersect in one point suggesting the ATP-competitive nature of inhibition of compound 10 on both EGFR and c-Met (Figure 2).
Figure 2.

Assessing the binding mode of compound 10 to recombinant (A) wt EGFR and (B) c-Met enzyme. Enzyme activity (Vmax) measurements were performed at various ATP and kinase inhibitor concentrations. The measured signal shifts (ΔS) were depicted on a Lineweaver–Burk double reciprocal plot.
All of the compounds showed time-dependent kinetics for inhibition of c-Met, which was proved by applying prolonged time of preincubation resulting in lower IC50 values similarly to XL-880 (foretinib), quinoline-based c-Met inhibitor.25 The IC50 values of all compounds were determined on clinically relevant NSCLC cell lines and on the NIH3T3 cell line, as nontumorous control. The NSCLC cell lines harbor various abnormalities in the EGFR signaling axis; therefore, the following different reference inhibitors were used: BMS-77760726 or crizotinib27 for H1993, erlotinib28 for HCC827, and afatinib29 for H1975 and A549. In accordance with their enzyme inhibitory profiles, 1 and 2 impressively reduced the viability of c-Met amplified H1993 cell line but showed a weaker effect against A549, HCC827, and H1975 cell lines containing EGFR or KRAS mutations. Furthermore, compounds 1 and 2 also diminished the viability of NIH3T3 cell line. Compounds 8–10 had weaker but more uniform effect on the four cell lines, without considerably inhibiting NIH3T3. Compound 10 was the most efficient among them on H1993 and on HCC827 cell lines, probably due to its more potent kinase inhibition profile. Cell viability inhibition of the H1975 cell line is probably also due to the c-Met inhibitory ability of 10.14 However, the effect on A549 cell line was weak, presumably because of its KRAS mutation beside the wt EGFR.30,31 To assess whether these compounds downregulate EGFR and c-Met kinase activation in living cells, we investigated downstream ERK-mediated MAPK and PI3K/AKT/mTOR signaling pathways and performed Western blot analysis on the tested cell lines. Compounds 1, 2, 8, and 9 inhibited c-Met phosphorylation but had no such effect on EGFR (data not shown). Compound 10 downregulated both c-Met and EGFR phosphorylation at 1 μM but abrogated downstream phosphorylation only on the c-Met inhibitor-sensitive H1993 cell line and not on the EGFR inhibitor-sensitive HCC827 cell line, probably due to its stronger effect on c-Met kinase activity than on EGFR (Figure 3).
Figure 3.
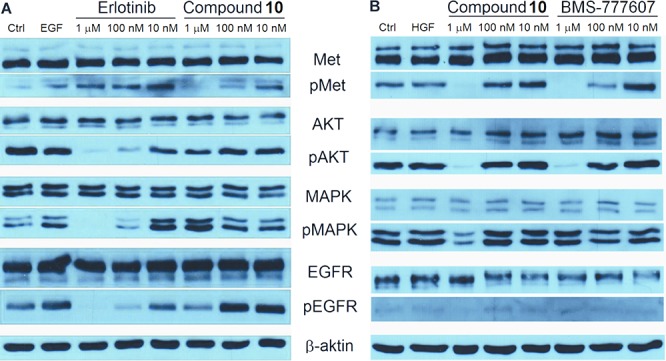
Western blot analysis of NSCLC cell lysates. (A) HCC827 cell line was treated with erlotinib and compound 10. (B) H1993 cell line was treated with BMS-777607 and compound 10.
The HCC827 and H1993 cell lines were exposed to control compounds and compound 10, which induced apoptosis on the HCC827 cell line almost as efficiently as erlotinib and afatinib, while none of the compounds had significant effect on H1993 cell line (Figure 4).
Figure 4.

Flow cytometry analysis of HCC827 and H1993 cell lines treated with erlotinib, crizotinib, afatinib, and compound 10. Data are means ± SD from at least 3 independent experiments. The significance of differences in the percentage of apoptosis was evaluated with unpaired 2-tailed Student’s t test. The * marks p < 0.05 versus the corresponding value for cells treated with DMSO.
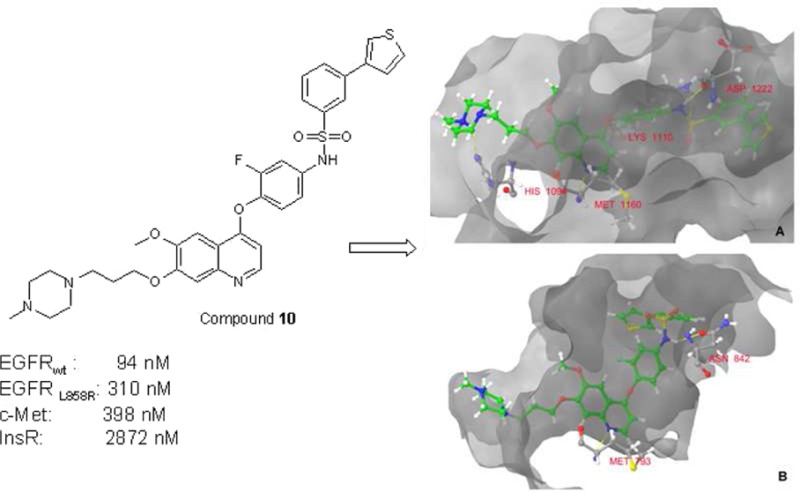
It has been reported that inhibition of c-Met leads to blocking of c-Met-triggered cell scattering in MDCK cells21,32 and DU145 cells,33 which is an excellent model for invasive growth and cell motility of tumor cells. Cell scattering of DU145 prostate cancer cells and MDCK cells were induced by HGF and treated by compound 10 to demonstrate the inhibitory effect of HGF-induced cell scattering (Supporting Information). In the case of DU145 cells, compound 10 showed to be less effective (IC50: 1.1 μM) than the reference compound BMS-777607 (IC50: 200 nM), which was used as a positive control due to its stronger c-Met inhibition. On MDCK cells, compound 10 induced cell death at 1 μM concentration, while at lower concentrations a low level of inhibition of cell scattering was observed. To assist further chemical modifications and to understand the mode of action, the EGFR/c-Met inhibitors were docked into the ATP binding site of both enzyme in silico using Protein Data Bank cocrystallization data (c-Met, 3LQ8; EGFR, 1XKK). By studying the predicted binding mode of the compounds on c-Met kinase, the following conclusions can be made: the quinoline N forms a hydrogen bond to Met1160 at the hinge region. The sulfonamide group interacts with the DFG motif at Asp1222 and with Lys1110 near the αC helix. The substituted phenyl, thiophene, or pyrazole groups insert into the small hydrophobic pocket near the DFG motif. The side chain 1-methyl-piperazine and the morpholine rings form other hydrogen bonds with the binding site improving the binding affinity of the compounds. In the case of the EGFR kinase, the quinoline nitrogen forms a hydrogen bond to Met793 at the hinge region and the sulfonamide group interacts with Asn842, which can be found near the DFG motif of the kinase (Figure 5).
Figure 5.
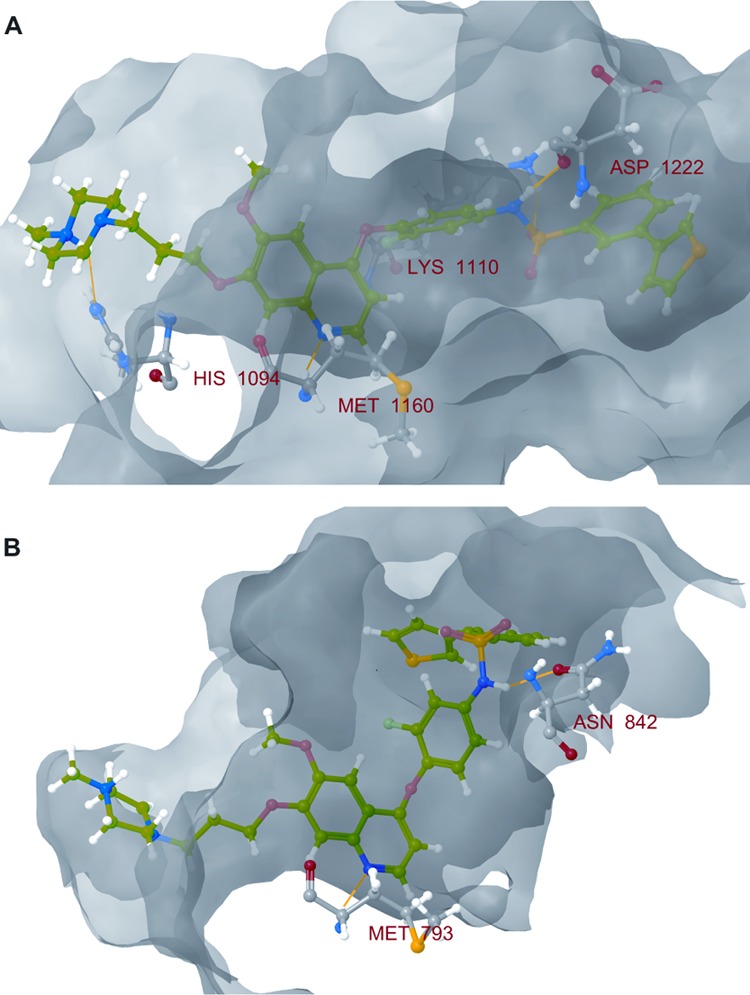
Predicted binding mode of compound 10 to (A) c-Met and (B) EGFR kinases. The formed hydrogen bonds are marked with yellow.
In conclusion, we have developed a novel N-[3-fluoro-4-[[6,7-disubstitued]-4-quinolinyl]oxy]phenyl]-biaryl-benzenesulfonamide-based multikinase inhibitor cotargeting EGFR and c-Met and characterized it in various biochemical assays. We found that compound 10 inhibited both EGFR and c-Met activity in nanomolar range in enzymatic assays and acts as a reversible, ATP-competitive inhibitor without considerably inhibiting InsR. Furthermore, compound 10 effectively inhibited cell viability on NSCLC cell lines A549, H1975, and especially HCC827 and H1993. We verified that this compound blocks phosphorylation of both enzymes at cellular level and induces apoptosis on HCC827 cell line comparable to reference compounds. On the cell motility model, inhibition of HGF-induced cell scattering was observed on the DU145 cell line at about 1 μM IC50. Compound 10, as a novel dual inhibitor of EGFR and c-Met, can be considered as a good starting point to seek more effective EGFR/c-Met inhibitors as drug candidates.
Acknowledgments
We are grateful to Dr. Lars Neumann for the technical assistance and to Dr. András Dancsó (EGIS Pharmaceuticals PLC, Budapest, Hungary) for providing help in molecular modeling studies.
Glossary
ABBREVIATIONS
- EGFR
epidermal growth factor receptor
- wt
wild-type
- c-Met
mesenchymal-epithelial transition factor
- NSCLC
non-small cell lung cancer
- HGF
hepatocyte growth factor
- RTKs
receptor tyrosine kinases
- TKI
tyrosine kinase inhibitor
- InsR
insulin receptor
- μW
microwave irradiation
- MAPK
microtubule-associated protein kinase
- PI3K
phosphatidylinositol-3-kinase
- AKT (PKB)
protein kinase B
- mTOR
mammalian target of rapamycin
Supporting Information Available
Experimental procedures and biological measuring methods for 1–15. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
○ B.S. and P.G. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work is supported by NKFP_07_A2-NANODRUG grant.
The authors declare no competing financial interest.
Author Status
△ T.V. and G.K. are shared last authors.
Supplementary Material
References
- Lynch T. J.; Adjei A. A.; Bunn P. A. Jr.; Eisen T. G.; Engelman J.; Goss G. D.; Haber D. A.; Heymach J. V.; Jänne P. A.; Johnson B. E.; Johnson D. H.; Lilenbaum R. C.; Meyerson M.; Sandler A. B.; Sequist L. V.; Settleman J.; Wong K. K.; Hart C. S. Summary statement: novel agents in the treatment of lung cancer: advances in epidermal growth factor receptor-targeted agents. Clin. Cancer Res. 2006, 12, 4365–4371. [DOI] [PubMed] [Google Scholar]
- Mendelsohn J.; Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene 2000, 19, 6550–6565. [DOI] [PubMed] [Google Scholar]
- Paez J. G.; Jänne P. A.; Lee J. C.; Tracy S.; Greulich H.; Gabriel S.; Herman P.; Kaye F. J.; Lindeman N.; Boggon T. J.; Naoki K.; Sasaki H.; Fujii Y.; Eck M. J.; Sellers W. R.; Johnson B. E.; Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [DOI] [PubMed] [Google Scholar]
- Sordella R.; Bell D. W.; Haber D. A.; Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [DOI] [PubMed] [Google Scholar]
- Lynch T. J.; Bell D. W.; Sordella R.; Gurubhagavatula S.; Okimoto R. A.; Brannigan B. W.; Harris P. L.; Haserlat S. M.; Supko J. G.; Haluska F. G.; Louis D. N.; Christiani D. C.; Settleman J.; Haber D. A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [DOI] [PubMed] [Google Scholar]
- Pao W.; Miller V. A.; Politi K. A.; Riely G. J.; Somwar R.; Zakowski M. F.; Kris M. G.; Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S.; Boggon T. J.; Dayaram T.; Jänne P. A.; Kocher O.; Meyerson M.; Johnson B. E.; Eck M. J.; Tenen D. G.; Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [DOI] [PubMed] [Google Scholar]
- Kosaka T.; Yatabe Y.; Endoh H.; Yoshida K.; Hida T.; Tsuboi M.; Tada H.; Kuwano H.; Mitsudomi T. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin. Cancer Res. 2006, 12, 5764–5769. [DOI] [PubMed] [Google Scholar]
- Balak M. N.; Gong Y.; Riely G. J.; Somwar R.; Li A. R.; Zakowski M. F.; Chiang A.; Yang G.; Ouerfelli O.; Kris M. G.; Ladanyi M.; Miller V. A.; Pao W. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [DOI] [PubMed] [Google Scholar]
- Looyenga B. D.; Cherni I.; Mackeigan J. P.; Weiss G. J. Tailoring tyrosine kinase inhibitors to fit the lung cancer genome. Transl. Oncol. 2011, 4, 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella M. C.; Comoglio P. M. HGF: a multifunctional growth factor controlling cell scattering. Int. J. Biochem. Cell Biol. 1999, 31, 1357–1362. [DOI] [PubMed] [Google Scholar]
- Engelman J. A.; Zejnullahu K.; Mitsudomi T.; Song Y.; Hyland C.; Park J. O.; Lindeman N.; Gale C. M.; Zhao X.; Christensen J.; Kosaka T.; Holmes A. J.; Rogers A. M.; Cappuzzo F.; Mok T.; Lee C.; Johnson B. E.; Cantley L. C.; Jänne P. A. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–43. [DOI] [PubMed] [Google Scholar]
- Bean J.; Brennan C.; Shih J. Y.; Riely G.; Viale A.; Wang L.; Chitale D.; Motoi N.; Szoke J.; Broderick S.; Balak M.; Chang W. C.; Yu C. J.; Gazdar A.; Pass H.; Rusch V.; Gerald W.; Huang S. F.; Yang P. C.; Miller V.; Ladanyi M.; Yang C. H.; Pao W. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20932–20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z.; Du R.; Jiang S.; Wu C.; Barkauskas D. S.; Richey J.; Molter J.; Lam M.; Flask C.; Gerson S.; Dowlati A.; Liu L.; Lee Z.; Halmos B.; Wang Y.; Kern J. A.; Ma P. C. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br. J. Cancer 2008, 99, 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. J.; Jiang Y. L.; Lin C. M.; Tsai S. C.; Peng S. F; Fushiya S.; Hour M. J.; Yang J. S. Dual inhibition of EGFR and c-Met kinase activation by MJ-56 reduces metastasis of HT29 human colorectal cancer cells. Int. J. Oncol. 2013, 43, 141–50. [DOI] [PubMed] [Google Scholar]
- Liu X.; Newton R. C.; Scherle A. P. Development of c-MET pathway inhibitors. Expert Opin. Investig. Drugs 2011, 20, 1225–1241. [DOI] [PubMed] [Google Scholar]
- Kao H. F.; Lin C. C.; Yang J. C. EGFR inhibitors as the first-line systemic treatment for advanced non-small-cell lung cancer. Future Oncol. 2013, 9, 991–1003. [DOI] [PubMed] [Google Scholar]
- Mollard A.; Warner S. L.; Call L. T.; Wade M. L.; Bearss J. J.; Verma A.; Sharma S.; Vankayalapati H.; Bearss D. J. Design, synthesis, and biological evaluation of a series of novel AXL kinase inhibitors. ACS Med. Chem. Lett. 2011, 212907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo N. D.; Bellon S. F.; Booker S. K.; Cheng Y.; Coxon A.; Dominguez C.; Fellows I.; Hoffman D.; Hungate R.; Kaplan-Lefko P.; Lee M. R.; Li C.; Liu L.; Rainbeau E.; Reider P. J.; Rex K.; Siegmund A.; Sun Y.; Tasker A. S.; Xi N.; Xu S.; Yang Y.; Zhang Y.; Burgess T. L.; Dussault I.; Kim T. S. Design, synthesis, and biological evaluation of potent c-Met inhibitors. J. Med. Chem. 2008, 51, 5766–79. [DOI] [PubMed] [Google Scholar]
- Li S.; Zhao Y.; Wang K.; Gao Y.; Han J.; Cui B.; Gong P. Discovery of novel 4-(2-fluorophenoxy)quinoline derivatives bearing 4-oxo-1,4-dihydrocinnoline-3-carboxamide moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. 2013, 21, 2843–2855. [DOI] [PubMed] [Google Scholar]
- Furlan A.; Colombo F.; Kover A.; Issaly N.; Tintori C.; Angeli L.; Leroux V.; Letard S.; Amat M.; Asses Y.; Maigret B.; Dubreuil P.; Botta M.; Dono R.; Bosch J.; Piccolo O.; Passarella D.; Maina F. Identification of new aminoacid amides containing the imidazo[2,1-b]benzothiazol-2-ylphenyl moiety as inhibitors of tumorigenesis by oncogenic Met signaling. Eur. J. Med. Chem. 2012, 47, 239–54. [DOI] [PubMed] [Google Scholar]
- Cai Z. W.; Wei D.; Schroeder G. M.; Cornelius L. A.; Kim K.; Chen X. T.; Schmidt R. J.; Williams D. K.; Tokarski J. S.; An Y.; Sack J. S.; Manne V.; Kamath A.; Zhang Y.; Marathe P.; Hunt J. T.; Lombardo L. J.; Fargnoli J.; Borzilleri R. M. Discovery of orally active pyrrolopyridine- and aminopyridine-based Met kinase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 3224–3229. [DOI] [PubMed] [Google Scholar]
- Li S.; Huang Q.; Liu Y.; Zhang X.; Liu S.; He C.; Gong P. Design, synthesis and antitumour activity of bisquinoline derivatives connected by 4-oxy-3-fluoroaniline moiety. Eur. J. Med. Chem. 2013, 64, 62–73. [DOI] [PubMed] [Google Scholar]
- Li J.; Wu N.; Tian Y.; Zhang J.; Wu S. Aminopyridyl/pyrazinyl spiro[indoline-3,4′-piperidine]-2-ones as highly selective and efficacious c-Met/ALK inhibitors. ACS Med. Chem. Lett. 2013, 4, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian F.; Engst S.; Yamaguchi K.; Yu P.; Won K. A.; Mock L.; Lou T.; Tan J.; Li C.; Tam D.; Lougheed J.; Yakes F. M.; Bentzien F.; Xu W.; Zaks T.; Wooster R.; Greshock J.; Joly A. H. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009, 69, 8009–8016. [DOI] [PubMed] [Google Scholar]
- Schroeder G. M.; An Y.; Cai Z. W.; Chen X. T.; Clark C.; Cornelius L. A.; Dai J.; Gullo-Brown J.; Gupta A.; Henley B.; Hunt J. T.; Jeyaseelan R.; Kamath A.; Kim K.; Lippy J.; Lombardo L. J.; Manne V.; Oppenheimer S.; Sack J. S.; Schmidt R. J.; Shen G.; Stefanski K.; Tokarski J. S.; Trainor G. L.; Wautlet B. S.; Wei D.; Williams D. K.; Zhang Y.; Zhang Y.; Fargnoli J.; Borzilleri R. M. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J. Med. Chem. 2009, 52, 1251–1254. [DOI] [PubMed] [Google Scholar]
- Timofeevski S. L.; McTigue M. A.; Ryan K.; Cui J.; Zou H. Y.; Zhu J. X.; Chau F.; Alton G.; Karlicek S.; Christensen J. G.; Murray B. W. Enzymatic characterization of c-Met receptor tyrosine kinase oncogenic mutants and kinetic studies with aminopyridine and triazolopyrazine inhibitors. Biochemistry 2009, 48, 5339–5349. [DOI] [PubMed] [Google Scholar]
- Moyer J. D.; Barbacci E. G.; Iwata K. K; Arnold L.; Boman B.; Cunningham A.; DiOrio C.; Doty J.; Morin M. J.; Moyer M. P.; Neveu M.; Pollack V. A.; Pustilnik L. R.; Reynolds M. M.; Sloan D.; Theleman A.; Miller P. Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res. 1997, 57, 4838–4848. [PubMed] [Google Scholar]
- Reid A.; Vidal L.; Shaw H.; de Bono J. Dual inhibition of ErbB1 (EGFR/HER1) and ErbB2 (HER2/neu). Eur. J. Cancer 2007, 43, 481–489. [DOI] [PubMed] [Google Scholar]
- Krypuy M.; Newnham G. M.; Thomas D. M.; Conron M.; Dobrovic A. High resolution melting analysis for the rapid and sensitive detection of mutations in clinical samples: KRAS codon 12 and 13 mutations in non-small cell lung cancer. BMC Cancer 2006, 6, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda K.; Tomizawa K.; Mitsudomi T. Biological and clinical significance of KRAS mutations in lung cancer: an oncogenic driver that contrasts with EGFR mutation. Cancer Metastasis Rev. 2010, 29, 49–60. [DOI] [PubMed] [Google Scholar]
- Patané S.; Pietrancosta N.; Hassani H.; Leroux V.; Maigret B.; Kraus J. L.; Dono R.; Maina F. A new Met inhibitory-scaffold identified by a focused forward chemical biological screen. Biochem. Biophys. Res. Commun. 2008, 375, 184–189. [DOI] [PubMed] [Google Scholar]
- Fram S. T.; Wells C. M.; Jones G. E. HGF-induced DU145 cell scatter assay. Methods Mol. Biol. 2011, 769, 31–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.