

Abstract
Here, we report on a mechanistic investigation based on DFT calculations and kinetic measures aimed at determining the energetics related to the cysteine nucleophilic attack on nitrile-carrying compounds. Activation energies were found to correlate well with experimental kinetic measures of reactivity with cysteine in phosphate buffer. The agreement between computations and experiments points to this DFT-based approach as a tool for predicting both nitrile reactivity toward cysteines and the toxicity of nitriles as electrophile agents.
Keywords: Cysteine, nucleophilic attack, DFT, nitrile reactivity
Nitriles are one of the most common chemical groups in nature. They are versatile synthetic intermediates and important compounds per se.1 They are endowed with rich chemistry and serve as precursors for various functional groups, such as amines, amidines, tetrazoles, aldehydes, amides, and other carboxy derivatives.2 They are also key motifs in numerous compounds of practical utility including dyes, herbicides, agrochemical, electronic materials, and drugs.3,4 In drug discovery, a deep interest in nitrile-containing pharmaceuticals is emerging, as shown by the high number (about 30) of nitrile-carrying drugs that are currently in use for a variety of pathological conditions.5 In addition, more than 20 nitrile-containing drug candidates are currently in clinical development. The emerging importance of nitrile in this field is due to its remarkable versatility. This is related to the short, polarized triple bond,6 which allows nitrile-containing molecules to have a large variety of different types of interactions. Several crystal structures of nitriles in complex with biological targets show the nitrile projecting into narrow clefts to make polar interactions or hydrogen bonds in sterically congested environments.7 They can also play a key role as hydrogen-bond acceptors. Several crystal structures show hydrogen bonding between the nitrile nitrogen and amino acids or water-mediated interactions with protein backbones.8 In other cases, the strong dipole facilitates polar interactions, in which the nitrile acts as a hydroxyl or carboxyl isostere.9 Most nitrile-containing pharmaceuticals are aromatics with aliphatic-, alkene-, and nitrogen-bound nitriles (cyanamides) being progressively less frequent. Very well-known examples of nitrile-containing pharmaceuticals are the inhibitors of aromatase, such as Anastrazole10 or Letrozole,11 used for the treatment of estrogen-dependent breast cancer.
Nitriles can react with serine or cysteine residues of proteases to afford an imidate or thioimidate covalent adduct, respectively.12 The use of a nitrile group as a warhead has gained considerable importance in covalent drug discovery,13 as this functional group is chemically less reactive than aldehydes. Several nitrile derivatives have been developed as reversible covalent modifiers of serine and/or cysteine proteases, including DPP-IV14 and cathepsin inhibitors15 for the treatment of type 2 diabetes and osteoporosis. In the first case, the inhibitors (e.g., vildagliptin16) covalently interact with the active site Ser610 residue, forming a reversible covalent imidate adduct. Similarly, a search for cathepsin K inhibitors led to a series of amidoacetonitrile inhibitors, in which the nitrile participates in a reversible, covalent interaction with the active site cysteine residue.17
There are three known chemical classes of nitrile-containing covalent inhibitors: (i) heteroaromatic nitriles, (ii) cyanamides, and (iii) amidoacetonitriles.5,18 Despite their different electrophilicity, all three classes can reversibly react with biological thiols (including glutathione and proteins) giving a thiomidate intermediate according to a chemical process resembling the Pinner reaction. Moreover, when the thiol group belongs to a cysteine with a free amino group, the readily formed thiomidate rapidly evolves into a thiazoline product (Scheme 1), whose chemical stability makes the overall reaction an irreversible process.18,19
Scheme 1. Nitrile-Containing Compounds Reacting with a Cysteine Residue.
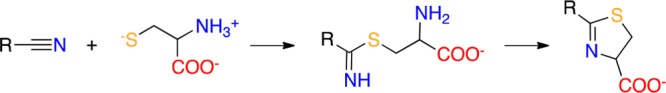
The nitriles react with cysteine residue forming a thioimidate intermediate that readily evolves into a thiazoline product.
Computational tools20 and/or experimental measures can be used to predict reactivity toward biological targets and toxicity toward off-targets. This is emerging as a key strategy in the discovery of novel nitrile-containing drugs.18,21,22 Oballa et al.18 calculated the theoretical reactivities of structurally diverse nitriles as the energy differences between the thioimidate adduct, a precursor thiol nucleophile, and the parent nitriles. In MacFaul et al.,21 the authors reported on an in vitro assay for assessing the reactivity of nitrile-containing compounds toward glutathione (GSH) and cysteine. In Ehmke et al.,22 the use of density functional theory (DFT) calculations provided relative reactivities of the nitriles and helped predict their biological affinity and cytotoxicity.
In both Oballa’s and MacFaul’s studies,18,21 reaction with free cysteine was used as an experimental model to probe nitrile reactivity. Actually, this system is only representative of reactions in biological environments where a thiolate attack is assisted by a proton transfer mechanism. This mechanism is found in many cysteine proteases, and particularly with Ntn-hydrolase,23 where a terminal cysteamine-like fragment acts both as a nucleophile and general acid.
Here, we present a combined computational and experimental study aimed at assessing the predictive power of a DFT-based tool (B3LYP/6-311++G(d,p)) in studying nitriles as covalent binders to cysteine residues. In previous studies, the electrophilicities were estimated from the formation enthalpies of the thioimidate adduct, as the product of the reaction between methanethiol and nitriles. Then, the calculated enthalpies were used for qualitative correlations with experimental kinetic data. Although these approaches could provide meaningful information, they were based on comparisons between thermodynamic and kinetic entities. In the present study, activation energy (Ea) values, rather than enthalpy differences, were compared to kinetic experimental measures. In particular, we investigated the reaction mechanism of thioimidate formation (Scheme 1) and compared theoretical and experimental results.
As a model system for the DFT calculations, we used cysteamine in its zwitterionc form (Figure 1A), which leads to a thioimidate as the product of the nucleophilic attack on a nitrile. All the simulations were carried out in implicit water. We first identified and characterized the transition state (TS) structures, and then we investigated the intrinsic reaction coordinate (IRC) pathways that lead to the reactants (R) and to the product (P). In this way, we calculated the Ea going from R to TS. In parallel, the experimental reactivity of the nitriles with a large excess of cysteine was studied by means of HPLC–UV and HPLC–ESI–MS measures, monitoring the decreasing concentration of the nitrile compounds as the reaction proceeded at 37 °C (see Supporting Information). The reactivity of a subset of nitriles was also measured with cysteamine and GSH by HPLC–UV and HPLC–ESI–MS/MS. These data are reported in the Supporting Information (Figures S1 and S2).
Figure 1.
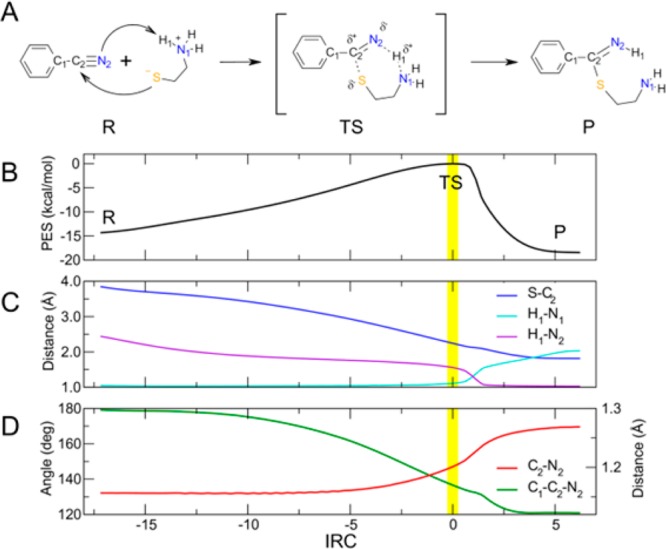
(A) Reaction mechanism using as a representative case benzonitrile. (B) PES as a function of IRC progression. (C,D) The progression along the IRC of the main geometrical features that change along the reaction.
Eleven different nitrile-carrying molecules were investigated comprising aliphatic, aromatic, and heteroaromatic species. In addition, three nitrile-containing pharmaceutical compounds (i.e., one drug, one clinical candidate, and one pharmacological tool) were studied using the same computational and experimental approaches.
In Figure 1, we report a schematic representation of the reaction here investigated (Figure 1A), the potential energy surface (PES) associated with the reaction (Figure 1B), and the evolution of some geometrical parameters as functions of the IRC (Figures 1C,D). Starting from the reactants, the sulfur atom of the cysteamine approached the carbon atom of the nitrile group: the distance S–C2 decreased, while H1 moved toward the nitrogen atom of the nitrile (see distance H1–N2 in Figure 1C). The hybridization of the nitrile carbon changed from sp to sp2 during the reaction. This could be deduced by observing the C2–N2 distance, which increased, whereas the C1–C2–N2 angle decreased (Figure 1D). The reaction occurred through a concerted synchronous mechanism. At the TS, the nucleophilic attack and the protonation happened simultaneously. Figure 1C shows that starting from the reactants the S–C2 and H1–N2 distances decreased along the IRC pathway.
As shown in Figure 2, the TS displayed a ring structure assuming a chair-like conformation. This chair-like conformation was remarkably similar to those previously reported for similar reactions in enzymatic environment.23−25 Furthermore, the concerted nature of the reaction mechanism here studied was also in good agreement with the amide hydrolysis catalyzed by classic cysteine hydrolases, such as papaine25 or cathepsin K.26
Figure 2.
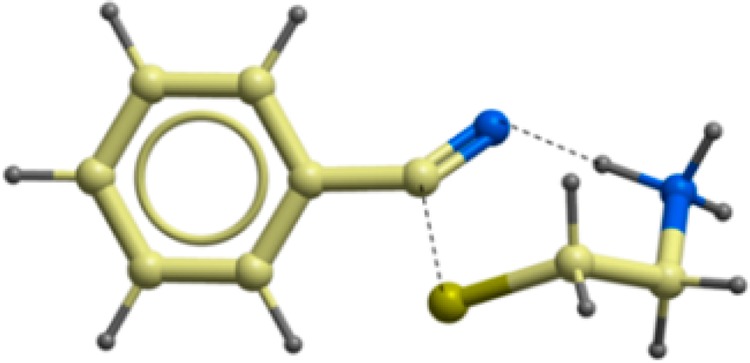
Transition state (TS) structure for benzonitrile 6 reacting with cysteamine. A chair-like structure composed of a 7-membered ring can be identified.
All the nitrile-carrying molecules here investigated (see Table 1) and the three nitrile-carrying pharmaceutical compounds (see Table 2) showed remarkably similar reaction mechanisms, which are reported in the Supporting Information. From our calculations, we estimated reactivity of nitriles from the Ea values, and we correlated these values to the experimental kinetic constants of the nitriles reacting with cysteine in phosphate buffer.
Table 1. Experimental t1/2 (min), Standard Deviation (SD), and Theoretical Activation Energy (Ea, kcal/mol) for Nitriles 1–11.
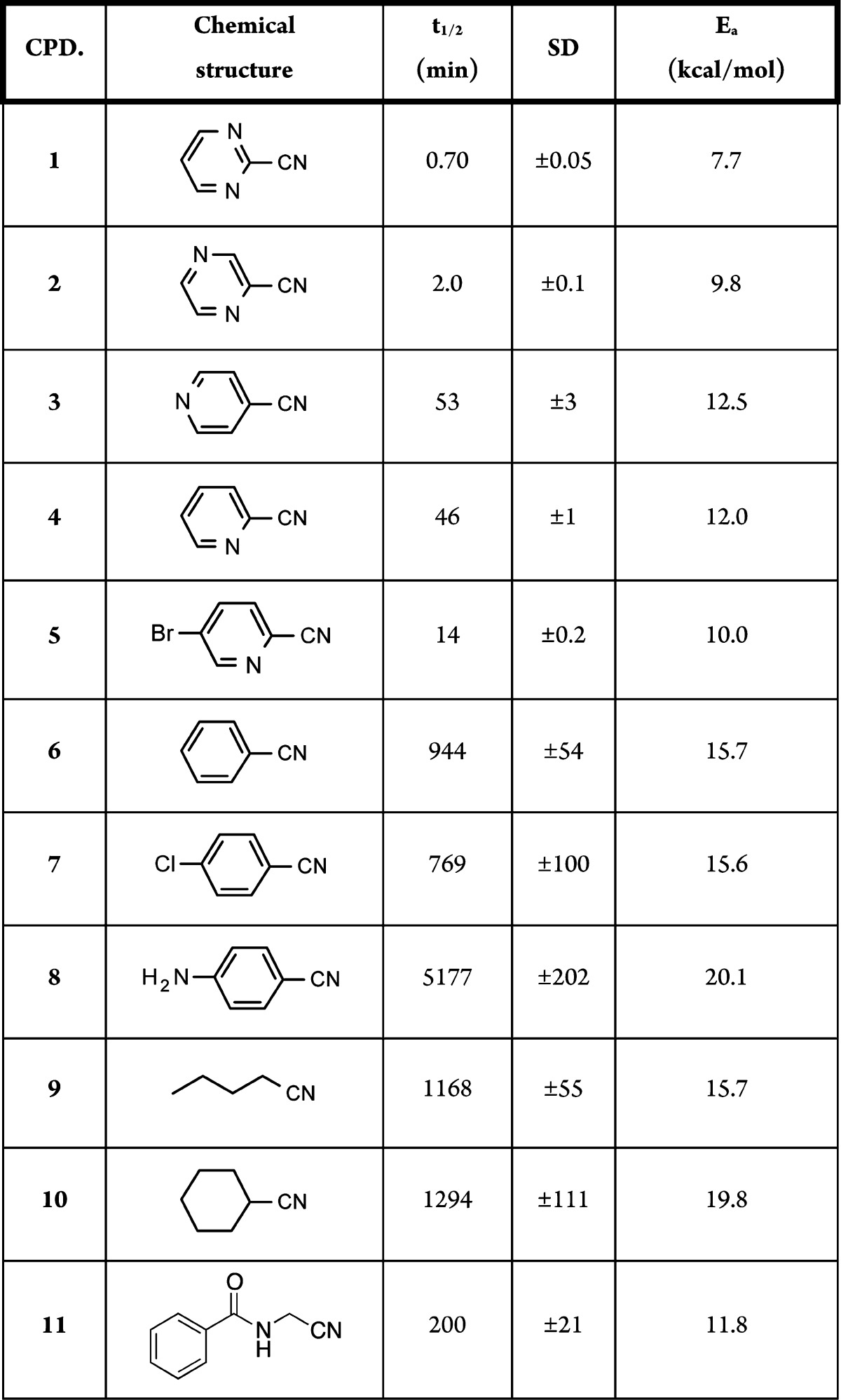
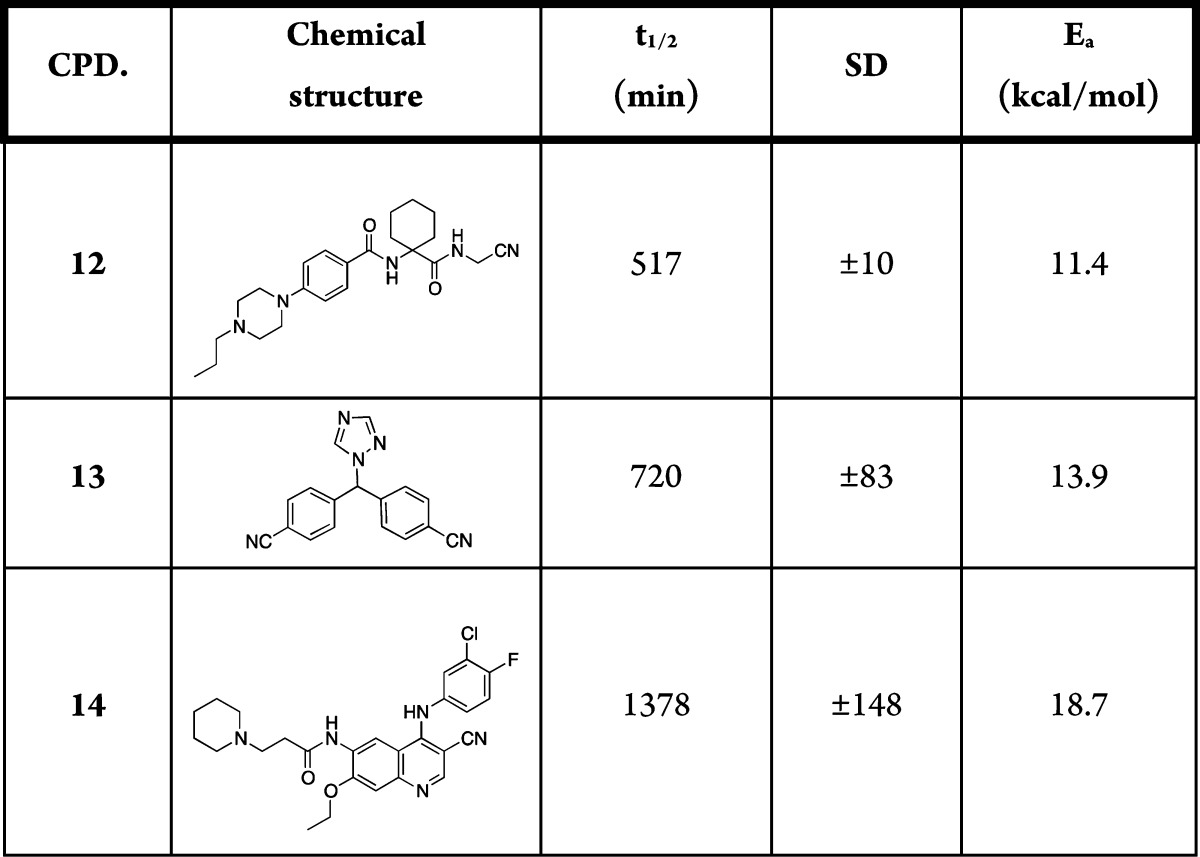
Table 2. Experimental t1/2 (min), Standard Deviation (SD), and Theoretical Activation Energy (Ea, kcal/mol) for Nitriles 12–14.
Among the most abundant ion species of cysteine at pH 7.4,27,28 the zwitterionic thiolate (depicted in Scheme 1) is probably the most reactive one. Under this hypothesis and considering the contribution of the carboxylate group as a constant, we used a cysteamine in the calculations as a simplified model of a cysteine.
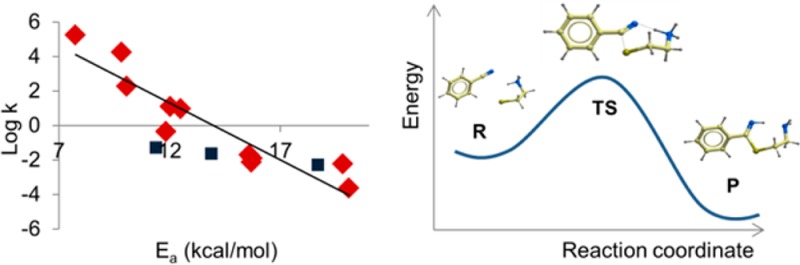
In Figure 3, we plotted the calculated Ea versus the experimental second-rate kinetic constants (k in M–1 min–1) reported on a log scale. As expected, pyrimidine, pyrazine, and pyridine nitriles (compounds 1–5 in Table 1) were highly reactive toward cysteine, due to the electron-withdrawing effect of the heteroaromatic rings linked to nitriles. The 2-cyanopyridine with a bromine atom in the para position (5) showed higher electrophilicity relative to the unsubstituted cyanopyridine; this can be attributed to the electron-withdrawing effect of the bromine atom. The benzonitrile (6) and the 4-chlorobenzonitrile (7) showed intermediate reactivities toward cysteine. When the benzonitrile was substituted with an electron donor amino group (8), the molecule was far less reactive; this was due to the electron donor effects of the substituent in position 4. The aliphatic nitriles (9 and 10) showed an electrophilicity, which was between that of the benzonitrile substituted in para with electron donor groups and the more electron-deficient heteroaromatic nitriles. Finally, the acetonitrile (11) was more reactive than benzonitriles 6–8 and less reactive than pyrimidine, pyrazine, and pyridine nitriles 1–5. Figure 3 also shows that computational data and experimental measures correlated well, with an R2 value of 0.86 (s = 1.14 and F = 54.1), considering compounds 1–11. Increasing the complexity of the structure, as with 12–14, decreased the predictive power of our DFT calculations. The reason for this limited predictivity is probably multifactorial as it depends both on some aproximations of B3LYP and on the use of static geometries, which do not take into account how costly it can be achieving the reactive conformation leading to the transition state.29
Figure 3.
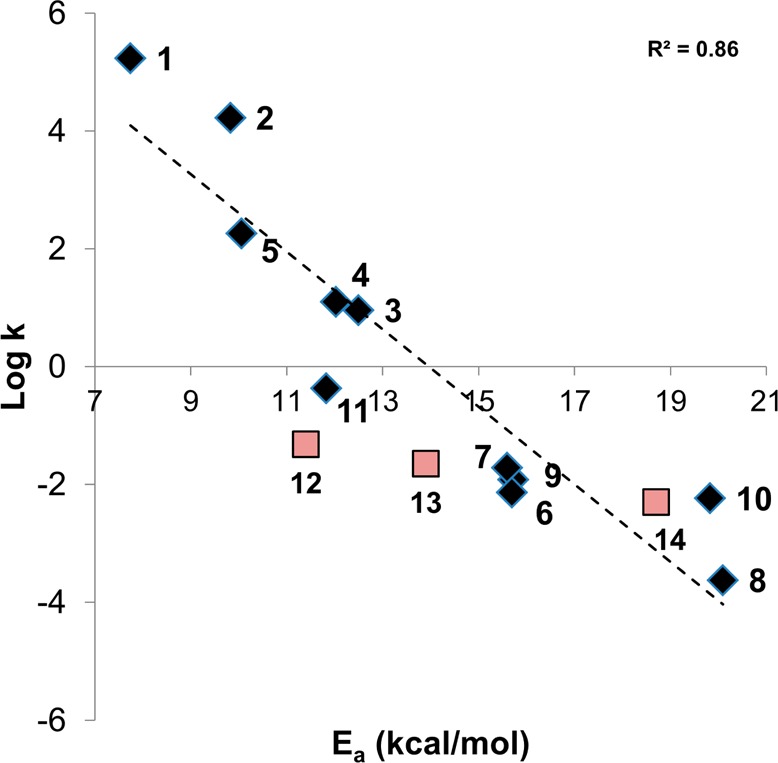
Correlation between the Ea calculated as the difference between the reactants and TSs and the experimental log(k). The black diamonds report the data from the compounds 1–11, while the red squares show the data for the three pharmaceutical compounds (12–14).
We finally performed additional single-point calculations at a higher level using the coupled-cluster theory with single and double excitations (CCSD). CCSD calculations provided the best correlation between theory and experiment (Figure S1, Supporting Information), with an R2 of 0.88 (s = 1.06 and F = 63.9). However, the computational cost of this method increases very rapidly with the number of atoms, which could restrict its application to quite small molecules. From a drug design perspective, DFT calculations, which are faster than CCSD ones, may be a good compromise between speed and accuracy.
The Ea values calculated by our approach can be considered as a relative scale expressing the propensity of a nitrile group, in a certain chemical environment, to react with thiol nucleophiles. Our computations are substantiated by experiments. They show that when the Ea is much less than 16 kcal/mol, nitriles can react with the cysteine on a small time scale, showing a reactivity similar to that of activated aromatic nitriles or aminoacetonitriles. These are generally considered to be covalent binders to biological targets. Conversely, when the Ea value approached or overcame 20 kcal/mol (e.g., compounds 8 and 10), the cysteine nucleophilic attack was extremely slow in our experimental conditions, with a half-life for nitrile disappearance that could be higher than 20 h, as observed for aliphatic nitriles or aromatic ones with conjugated electron-donating groups. The present DFT-based tool and the threshold value here reported could be used to predict how promptly a nitrile will react with a cysteine, leading to a covalent adduct. This could be extremely helpful in covalent drug design and for predicting possible off-target liabilities for nitrile-carrying drug candidates.
Acknowledgments
Dr. Philip MacFaul (AstraZeneca) is kindly acknowledged for providing compound 12 (Balicatib). The authors acknowledge the IIT Platform “Computation” for computational resources.
Supporting Information Available
Computational details, experimental details, geometry of all the stationary points investigated, and correlation plots between log(k) and Ea calculated at different levels of theory. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Patai S.; Rappoport Z. J.. The Chemistry of Triple-Bonded Functional Groups; Wiley & Sons: Chichester, U.K., 1983. [Google Scholar]
- Larock R. C.Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2nd ed.; VHC: New York, 1989. [Google Scholar]
- Fleming F. F.; Wang Q. Unsaturated nitriles: conjugate additions of carbon nucleophiles to a recalcitrant class of acceptors. Chem. Rev 2003, 103, 2035–2077. [DOI] [PubMed] [Google Scholar]
- Miller J. S.; Manson J. L. Designer magnets containing cyanides and nitriles. Acc. Chem. Res. 2001, 34, 563–570. [DOI] [PubMed] [Google Scholar]
- Fleming F. F.; Yao L.; Ravikumar P. C.; Funk L.; Shook B. C. Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010, 53, 7902–79 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Questel J.-Y.; Berthelot M.; Laurence C. Hydrogen-bond acceptor properties of nitriles: a combined crystallographic and ab initio theoretical investigation. J. Phys. Org. Chem. 2000, 13, 347–358. [Google Scholar]
- Laurence C.; Brameld K. A.; Graton J.; Le Questel J. Y.; Renault E. The pKBHX Database: Toward a Better Understanding of Hydrogen-Bond Basicity for Medicinal Chemists. J. Med. Chem. 2009, 52, 4073–4086. [DOI] [PubMed] [Google Scholar]
- Teno N.; Miyake T.; Ehara T.; Irie O.; Sakaki J.; Ohmori O.; Gunji H.; Matsuura N.; Masuya K.; Hitomi Y.; Nonomura K.; Horiuchi M.; Gohda K.; Iwasaki A.; Umemura I.; Tada S.; Kometani M.; Iwasaki G.; Cowan-Jacob S. W.; Missbach M.; Lattmann R.; Betschart C. Novel scaffold for cathepsin K inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6096–6100. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Duraiswami C.; Madauss K. P.; Tran T. B.; Williams S. P.; Deng S. J.; Graybill T. L.; Hammond M.; Jones D. G.; Grygielko E. T.; Bray J. D.; Thompson S. K. 2-Amino-9-aryl-3-cyano-4-methyl-7-oxo-6,7,8,9 tetrahydropyrido[2′,3′:4,5]thieno[2,3-b]pyridine derivatives as selective progesterone receptor agonists. Bioorg. Med. Chem. Lett. 2009, 19, 4916–4919. [DOI] [PubMed] [Google Scholar]
- Milani M.; Jha G.; Potter D. A. Anastrozole use in early stage breast cancer of post-menopausal women. Clin. Med. Ther. 2009, 1, 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar A. S. The discovery and mechanism of action of letrozole. Breast Cancer Res. Treat. 2007, 105Suppl 17–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour E.; Storer A. C.; Menard R. Peptide aldehydes and nitriles as transition state analog inhibitors of cysteine proteases. Biochemistry 1995, 34, 9136–9143. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Lohr F.; Furtmann N.; Klas J.; Gutschow M. Structural optimization of azadipeptide nitriles strongly increases association rates and allows the development of selective cathepsin inhibitors. J. Med. Chem. 2011, 54, 396–400. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Stirnberg M.; Sisay M. T.; Gutschow M. Development of nitrile-based peptidic inhibitors of cysteine cathepsins. Curr. Top. Med. Chem. 2010, 10, 294–322. [DOI] [PubMed] [Google Scholar]
- Mathieu C.; Degrande E. Vildagliptin: a new oral treatment for type 2 diabetes mellitus. Vasc. Health Risk Manag. 2008, 4, 1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. S.; Deschenes D.; Desmarais S.; Falgueyret J. P.; Gauthier J. Y.; Kimmel D. B.; Leger S.; Masse F.; McGrath M. E.; McKay D. J.; Percival M. D.; Riendeau D.; Rodan S. B.; Therien M.; Truong V. L.; Wesolowski G.; Zamboni R.; Black W. C. Identification of a potent and selective non-basic cathepsin K inhibitor. Bioorg. Med. Chem. Lett. 2006, 16, 1985–1989. [DOI] [PubMed] [Google Scholar]
- Oballa R. M.; Truchon J. F.; Bayly C. I.; Chauret N.; Day S.; Crane S.; Berthelette C. A generally applicable method for assessing the electrophilicity and reactivity of diverse nitrile-containing compounds. Bioorg. Med. Chem. Lett. 2007, 17, 998–1002. [DOI] [PubMed] [Google Scholar]
- Gauthier J. Y.; Chauret N.; Cromlish W.; Desmarais S.; Duong le T.; Falgueyret J. P.; Kimmel D. B.; Lamontagne S.; Leger S.; LeRiche T.; Li C. S.; Masse F.; McKay D. J.; Nicoll-Griffith D. A.; Oballa R. M.; Palmer J. T.; Percival M. D.; Riendeau D.; Robichaud J.; Rodan G. A.; Rodan S. B.; Seto C.; Therien M.; Truong V. L.; Venuti M. C.; Wesolowski G.; Young R. N.; Zamboni R.; Black W. C. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. [DOI] [PubMed] [Google Scholar]
- De Vivo M. Bridging quantum mechanics and structure-based drug design. Front. Biosci. 2011, 16, 1619–1633. [DOI] [PubMed] [Google Scholar]
- MacFaul P. A.; Morley A. D.; Crawford J. A simple in vitro assay for assessing the reactivity of nitrile containing compounds. J. Bioorg. Med. Chem. Lett. 2009, 19, 1136–1138. [DOI] [PubMed] [Google Scholar]
- Ehmke V.; Quinsaat J. E.; Rivera-Fuentes P.; Heindl C.; Freymond C.; Rottmann M.; Brun R.; Schirmeister T.; Diederich F. Tuning and predicting biological affinity: aryl nitriles as cysteine protease inhibitors. Org. Biomol. Chem. 2012, 10, 5764–5768. [DOI] [PubMed] [Google Scholar]
- Lodola A.; Branduardi D.; De Vivo M.; Capoferri L.; Mor M.; Piomelli D.; Cavalli A. A catalytic mechanism for cysteine N-terminal nucleophile hydrolases, as revealed by free energy simulations. PLoS One 2012, 7, e32397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perakyla M.; Rouvinen J. A serine protease-like mechanism with an N-terminal threonine and substrate-assisted catalysis. Chemistry 1996, 2, 1548–1551. [Google Scholar]
- Harrison M. J.; Burton N. A.; Hillier I. H. Catalytic mechanism of the enzyme papain: predictions with a hybrid quantum mechanical/molecular mechanical potential. J. Am. Chem. Soc. 1996, 119, 12285–12291. [Google Scholar]
- Ma S.; Devi-Kesavan L. S.; Gao J. Molecular dynamics simulations of the catalytic pathway of a cysteine protease: a combined QM/MM study of human cathepsin K. J. Am. Chem. Soc. 2007, 129, 13633–13645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch R. E.; Benesch R. The acid strenght of the −SH group in cysteine and related compounds. J. Am. Chem. Soc. 1955, 77, 5877–5881. [Google Scholar]
- Clement G. E.; Hartz T. P. Determination of microscopic ionization constants of cysteine. J. Chem. Educ. 1971, 48, 395–397. [DOI] [PubMed] [Google Scholar]
- Lodola A.; Sirirak J.; Fey N.; Rivara S.; Mor M.; Mulholland A. J. Structural fluctuations in enzyme-catalyzed reactions: determinants of reactivity in fatty acid amide hydrolase from multivariate statistical analysis of quantum mechanics/molecular mechanics paths. J. Chem. Theory Comput. 2010, 6, 2948–2960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.