

Abstract
LSP1-2111 is a group III metabotropic glutamate receptor agonist with preference toward the mGlu4 receptor subtype. This compound has been extensively used as a tool to explore the pharmacology of mGlu4 receptor activation in preclinical animal behavioral models. However, the blood–brain barrier penetration of this amino acid derivative has never been studied. We report studies on the central nervous system (CNS) disposition of LSP1-2111 using quantitative microdialysis in rat. Significant unbound concentrations of the drug relative to its in vitro binding affinity and functional potency were established in extracellular fluid (ECF). These findings support the use of LSP1-2111 to study the CNS pharmacology of mGlu4 receptor activation through orthosteric agonist mechanisms.
Keywords: Metabotropic glutamate receptors, G protein-coupled receptors, mGlu4 receptor, orthosteric agonist
The investigation of potential therapeutic indications of metabotropic glutamate 4 (mGlu4) receptor activation is an active area of drug discovery research.1 The selective activation of the mGlu4 receptor can be achieved using two different molecular mechanisms: orthosteric agonists (competing with l-glutamate, 1; see Chart 1) or noncompetitive positive allosteric modulators (PAMs).2
Chart 1. Chemical Structures of Selected mGlu4 Receptor Orthosteric Agonists (1–4), Marketed CNS Drugs (5–10), and mGlu2/3 Receptor Agonists which Reached Clinical Trials (11–14) Containing Amino Acid Functionality.

A number of mGlu receptor agonists with selectivity for Group III (mGlu4, 6, 7, and 8 receptors) versus Group I (mGlu1 and 5 receptors) and Group II (mGlu2 and 3 receptors) have been reported. From a drug design perspective, L-AP4 (2) may be considered as a starting lead compound toward the discovery of mGlu4 receptor agonists with improved subtype selectivity. LSP1-2111 [3; (2S)-2-amino-4-(hydroxy(hydroxy(4-hydroxy-3-methoxy-5-nitrophenyl)methyl)phosphoryl)butanoic acid, used as a 1:1 mixture of equipotent diastereoisomers at the benzylic position] is a preferential agonist of the mGlu4 and mGlu6 receptors, where the terminal phosphonic acid fragment in 2 is replaced by a hydroxymethyl-phosphinic acid pharmacophore, with an extra substituted aryl group.3,4 While highly selective (>100-fold) against Group I and II mGlu receptors, LSP1-2111 (3) has 1-, 25-, and 30-fold preference over mGlu6, mGlu7, and mGlu8 receptors, respectively.5 Recently, the discovery of a highly selective mGlu4 agonist was reported, known as LSP4–2022 (4, also obtained as a 1:1 mixture of diastereoisomers). This shows an improved selectivity profile of 40-, 100-, and 300-fold versus the mGlu6, 7, and 8 receptors, respectively.6
The interest of a number of drug discovery organizations, including our group, has focused in the PAM mechanism.1,2,7 However, recent reports prompted us to explore the orthosteric agonist mechanism. Specifically, preclinical pharmacology studies with LSP1-2111 reported efficacy using in vivo rodent models of Parkinson’s disease,8 anxiety,9 and psychosis.10,11 LSP1-2111 was also efficacious in acquisition of fear learning and memory models.12 These results in multiple animal models are both compelling and provocative. To our best knowledge, there are no reports of the concentrations of LSP1-2111 in tissues relevant to central pharmacological actions (plasma, brain, cerebrospinal fluid (CSF), or brain extracellular fluid (ECF)) in any species, in particular rat. This led to an investigation of central drug disposition and pharmacokinetics of LSP1-2111 following systemic administration in rat, to examine whether the compound is able to cross the blood–brain barrier (BBB) and achieve relevant exposures at the receptor location.13,14
Strategies to establish BBB permeability for typical (e.g., lipophilic) CNS drugs have changed over the past decade. Prior focus on total brain-to-plasma ratios has shifted to using parameters such as unbound drug concentration or unbound brain-to-plasma ratios during the lead optimization of CNS drugs.15 The compounds are often of lipophilic nature, show high levels of nonspecific binding to matrix components, and cross membranes through transcellular mechanisms. In general, an aim of the CNS drug design process is to avoid significant interactions with efflux transporters. However, the physicochemical characteristics of amino acids such as LSP1-2111 (Table 1) are markedly different from those for a typical CNS drug, which impacts their systemic disposition attributes. These compounds show low cell membrane passive permeability through the transcellular mechanism, which may lead to unbound brain-to-plasma ratios deviating from unity, as the free drug hypothesis would predict.16 They remain largely in the vascular compartment upon peripheral dosing, and in the brain they are mainly distributed in the extracellular space. They are mostly distributed as unbound in plasma and brain parenchyma. Therefore, we felt a different strategy was required to establish BBB penetration than that typically used for lipophilic drugs.
Table 1. In Vitro and in Silico Attributes of LSP1-2111 (3).
| h.mGlu4 EC50 (FLIPR) | 1.5 μM |
| h.mGlu4 Emax (FLIPR) | 85% |
| h.mGlu4 IC50 ([3H]-L-AP4 binding) | 8.6 μM |
| molecular weight | 364 amu |
| LogD7.4 | –0.7 |
| cLogP | –2.6 |
| polar surface area | 196 Å2 |
| aqueous solubility pH 7.4 | >1.8 mg/mL |
| pKa | 11.7; 6.6; 2.0 |
| passive permeability PAPP (PAMPA) | <0.1 × 10–6 cm/s |
| MDCK/MDR1 permeability PAPP | A → B = 0.2 × 10–6 cm/s |
| B → A = 0.5 × 10–6 cm/s | |
| human plasma unbound fraction, fu | 0.73 |
| rat or mouse plasma unbound fraction, fu | >0.99 |
| rat or mouse brain homogenate unbound fraction, UBBR | >0.99 |
| rat microsomal CLinta | 3.1 mL/min (low) |
| human microsomal CLintb | 0.1 L/min (low) |
| human hERG Ionworksc | 14% inhibition at 30 μM |
| cytotoxicity assayd | score 1 (nontoxic) |
| reactive oxygen species assayd | score 1 (nontoxic) |
| mitochondrial membrane potential assayd | nontoxic |
| broad selectivity screene | no cross-reactivity observed at 10 μM |
It is worth noting that a number of amino acid derivatives believed to act directly at brain receptors are used in the clinic for a number of CNS indications; for example, levodopa (5), baclofen (6), gabapentin (7), pregabalin (8), vigabatrin (9), and α-methyltyrosine (10). In addition, related amino acid mGlu2/3 agonists LY354740 (11) and LY404039 (12), as their corresponding prodrugs LY544344 (13) and LY2140023 (14), have reached the clinical stage. Our plan was to establish the concentration–time profile of LSP1-2111 in brain ECF using quantitative microdialysis techniques. This would measure the ability of LSP1-2111 to cross the BBB and access the orthosteric binding site on the extracellular space of the cell membrane-bound mGlu4 receptor.1,2
Prior to performing microanalysis studies, we characterized LSP1-2111 in terms of its physicochemical and in vitro ADMET profile (Table 1). Consistent with its hydrophilic nature, the experimental LogD7.4 (−0.7) and the in silico cLogP (−2.6) are low.17 The polar surface area (196 Å2) is well above the typical values considered optimum for lipophilic CNS drugs.18 The kinetic aqueous solubility measured in an assay developed and optimized for evaluating lipophilic compounds exceeded the detection limit (>800 μM). The thermodynamic solubility was high as well (>1.8 mg/mL). Consistent with its highly polar nature, the transcellular permeability was determined to be low by two different measures: a PAMPA assay and the MDCK/MDR1 assay (PAPP < 0.5 × 10–6 cm/s in both cases). The PAPP ratio B → A/A →B = 2.5 might suggest LSP1-2111 is a moderate P-glycoprotein (P-gp) substrate; albeit, these permeability values are very low and within experimental error. Human, rat, and mouse plasma unbound fractions are high (fu = 0.73, >0.99, and >0.99, respectively). Rat and mouse brain homogenate unbound fraction (>0.99 in both cases) indicate this compound has very low nonspecific binding to brain tissue. Both rat and human microsomal intrinsic clearance (CLint) are low, suggesting LSP1-2111 is poorly metabolized by hepatic cytochrome P450 enzymes. Standard in vitro toxicology assays indicated a rather safe profile, including low potential for hERG channel inhibition.19 Lastly, a broad binding screen against 70 GPCRs, ion channels, and enzymes as well as a functional screen toward 56 GPCRs demonstrated a highly selective cross-reactivity profile for LSP1-2111.20,21
We then determined the distribution characteristics of LSP1-2111 within CNS, using in vivo microdialysis to generate a time course profile of extracellular space fluid concentrations using a MetaQuant dialysis probe (Figure 1). The method follows the principle that microdialysis extraction becomes quantitative (i.e., concentration recovery is close to 100%) when the dialysis flow rate is decreased from the usual rates of 1.5–2 μL/min down to 0.1 μL/min. In this study, in vitro recovery of the probes was 96% under similar low flow rate conditions.22
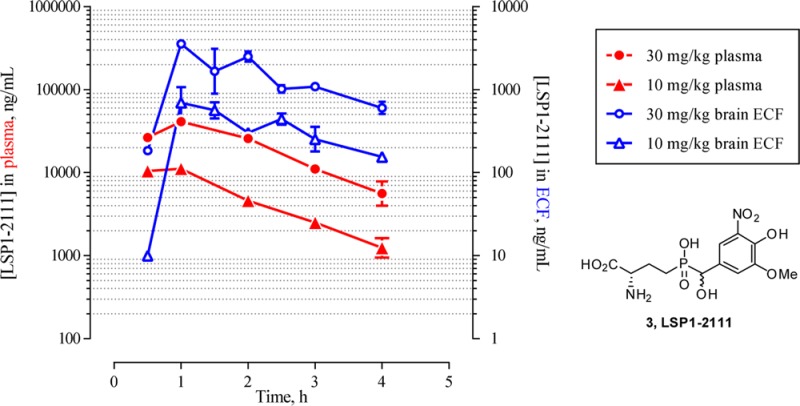
Figure 1.
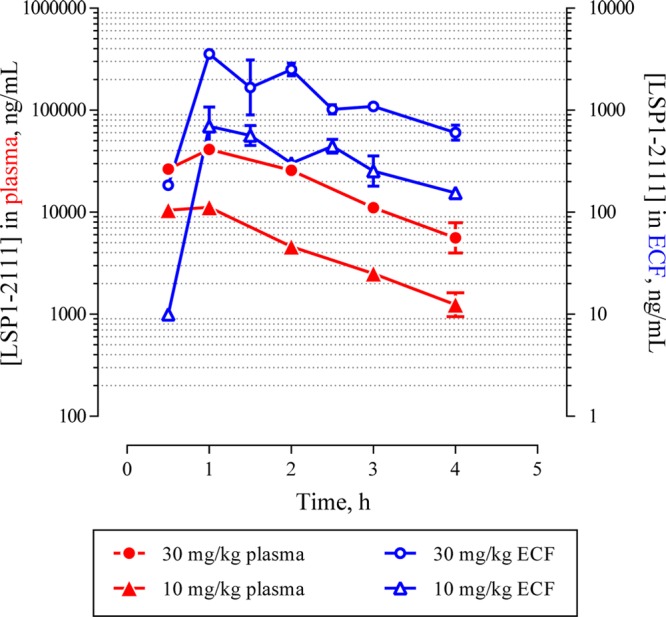
Rat plasma and ECF dialysate concentration time profile of LSP1-2111 following subcutaneous (SC) administration at 10 and 30 mg/kg. The ECF concentration values have been corrected for recovery loss based on the 96% in vivo probe recovery under the same flow rate.23
In the behavioral studies reported in the literature, LSP1-2111 was dosed via intraperitoneal (IP) administration.8−12 In our hands, preliminary work comparing brain and plasma exposures in rat upon IP or subcutaneous (SC) dosing demonstrated very similar average concentration values of LSP1-2111, but with a significantly higher inter-animal variability in the IP route (Supporting Information, Figure S1). Thus, we chose the SC route of administration for all our in vivo brain penetration studies with LSP1-2111.
Plasma concentrations in the microdialysis samples were dose-proportional and consistent with those found in the preliminary exposure studies. They reached maximum values of 11 ± 1 μg/mL (31 μM) and 41 ± 7 μg/mL (114 μM) at 10 and 30 mg/kg, respectively, 60 min after subcutaneous dose. ECF concentrations were dose-proportional, reaching values of up to 0.7 ± 0.4 μg/mL (1.8 μM) and 3.5 μg/mL (9.8 μM) at 10 and 30 mg/kg, respectively. These concentrations are commensurate with the in vitro binding affinity and functional potency of LSP1-2111 (Table 1 and those reported in the literature).3,7
Further insight into the CNS disposition of LSP1-2111 was gained upon studying its distribution into CSF and brain tissue after systemic administration. Tissue concentrations were measured in rats after dosing LSP1-2111 (10 mg/kg, SC) formulated in saline at pH 7.4. Results from a 6-h time course study are shown in Figure 2.
Figure 2.
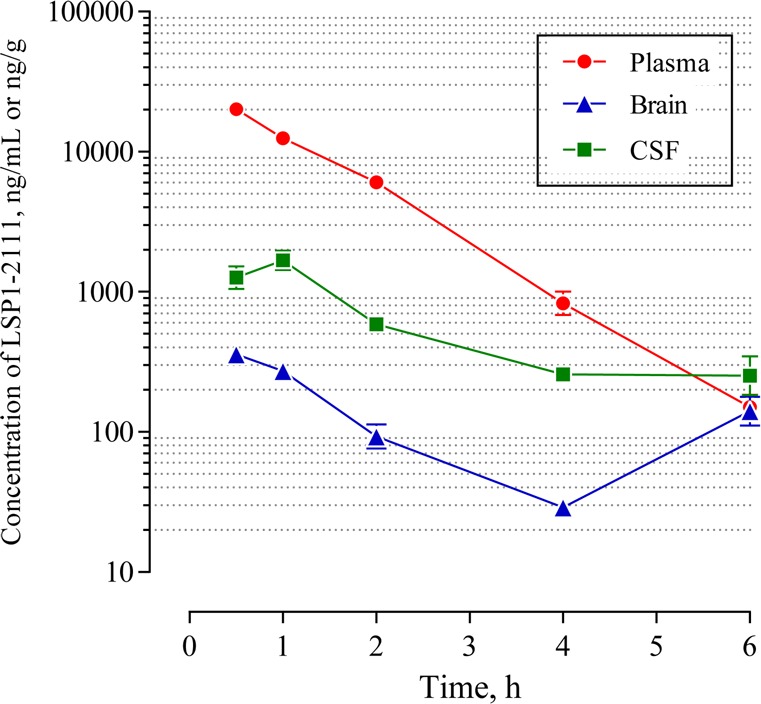
Plasma, brain, and CSF concentration time profile of LSP1-2111 upon subcutaneous (SC) administration at 10 mg/kg (N = 4). Only standard error bars larger than marker size are shown.
Plasma concentrations decreased with a t1/2 of 20 min. The average plasma concentrations of LSP1-2111 during the 0.5–6 h time interval decreased from 20.1 ± 3.7 μg/mL to 0.15 ± 0.05 μg/mL, with a maximum unbound plasma concentration of 55.2 μM at 0.5 h. Plasma, brain, and CSF AUC0–6 values were 30 μg × h/mL, 0.7 μg × h/g, and 3.5 μg × h/mL, respectively.
The average CSF concentrations for LSP1-2111 also declined from 1.7 ± 0.5 μg/mL (4.6 μM) to 0.3 ± 0.1 μg/mL (0.7 μM) over the 6 h period following SC dose administration. Since protein content in CSF is well below that of plasma, these concentrations essentially represent the free drug. Thus, the maximum CSF concentration of LSP1-2111 (4.6 μM) is ca. 2-fold below its binding affinity, ca. 2-fold above its functional EC50 (Table 1), and ca. 2.5-fold the corresponding ECF concentration (1.8 μM) at 1 h for the 10 mg/kg dose. The CSF AUC0–6 is approximately 12% of the corresponding plasma AUC0–6.
The average brain homogenate concentrations of LSP1-2111 range between 0.36 ± 0.07 μg/g (1 μM) at 30 min and 0.03 ± 0.01 μg/g (0.08 μM) at 4 h. This essentially follows the rate of change in plasma concentrations over the same period. The 6 h drug concentration is 0.14 ± 0.05 μg/g (0.4 μM). Based on AUC0–6, the brain-to-plasma ratio for LSP1-2111 is 2.4%. This low value is in agreement with previous reports for related compounds such as 11 and 13.24 As a general practice in our experimental protocols, brain tissue was not perfused to eliminate capillary blood prior to homogenization in the measurement of LSP1-2111 concentration. Thus, the concentrations measured in the brain tissue are mostly arising from LSP1-2111 in blood contained in brain capillaries, outside of the CNS structure (1.9% wet brain weight).25 This precludes a clear determination of BBB penetration based on brain concentrations of LSP1-2111, especially when taken into consideration experimental variability.
It is noteworthy that despite their low passive permeability characteristics, sporadic reports have disclosed that similar amino acid compounds are orally absorbed in preclinical species, presumably facilitated by active transport processes.26 Thus, we decided to explore whether intestinal absorption of LSP1-2111 could be facilitated by putative transporters. Results from an in vivo rat pharmacokinetic study in Sprague–Dawley rats are shown in Figure 3, and key parameters are listed in Table 2. Plasma clearance is low (6.2 mL/min × kg) compared with the characteristic hepatic blood flow of 75 mL/min × kg. The volume of distribution at steady state is very low (Vss = 0.1 L/kg), indicating the majority of the compound dosed remains in the vascular compartment. Unfortunately, oral bioavailability was poor (0.8%), and Cmax was 92 ng/mL (253 nM unbound drug concentration), showing that very low systemic exposure was obtained following the oral route of administration. Plasma t1/2 was short (0.3 h), as a reflection of a very low Vss.
Figure 3.
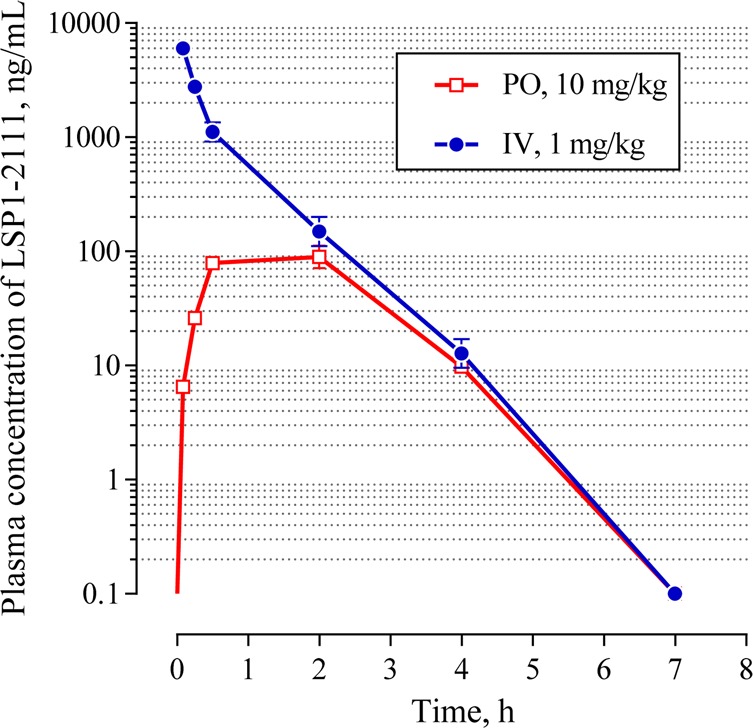
Plasma concentration time profile of LSP1-2111 after oral administration in SD rats (N = 2). Only standard error bars larger than marker size are shown.
Table 2. Summary of Rat Pharmacokinetic Parameters for LSP1-2111 Generated Using Noncompartmental Analysis in WinNonlin 5.2a.
| parameter | units | intravenous (IV)b | oral (PO)b |
|---|---|---|---|
| dose | mg/kg | 1 | 10 |
| t1/2 | h | 0.3 | |
| Cmax | ng/mL | 92 | |
| tmax | h | 1.4 | |
| Clp | mL/min × kg | 6.2 | |
| Vss | L/kg | 0.1 | |
| AUCinf | μg × h/mL | 2.8 | 0.2 |
| F | % | 0.8 |
LSP1-2111 (3) was dosed as a solution in saline buffered to pH 7.4, at a dose volume of 5 mL/kg.
Mean values (N = 2).
In summary, we report the use of quantitative microdialysis for the determination of brain ECF concentrations for LSP1-2111, an amino acid tool compound extensively used to study behavioral pharmacology of mGlu4 receptor activation. The ECF concentrations achieved at the doses tested, which partly overlap with those showing efficacy in rat models, are commensurate with the compound’s mGlu4 receptor binding affinity and functional EC50 values. Due to the particular physicochemical and ADMET attributes of highly polar amino acids, the total brain-to-plasma ratio does not necessarily provide an appropriate measure of brain penetration. The oral bioavailability of LSP1-2111 in rat is low, suggesting the absence of intestinal transporters to facilitate its absorption. LSP1-2111 is a useful tool compound to explore the potential of mGlu4 agonists as therapies for CNS and peripheral disorders. Thus, selective orthosteric agonists should provide valuable options to complement the ongoing efforts in potentiating the mGlu4 receptor via positive allosteric modulation.
Acknowledgments
We are grateful to Dr. Stevin Zorn and Dr. Klaus Bæk Simonsen for support of this work. We thank the reviewers of this manuscript for their valuable criticism.
Glossary
Abbreviations
- ADMET
absorption-distribution-metabolism-elimination-toxicity
- PAM
positive allosteric modulator
- CSF
cerebrospinal fluid
- ECF
extracellular fluid
- CNS
central nervous system
- SC
subcutaneous
- IP
intraperitoneal
- P450
cytochrome P450
- GPCR
G protein-coupled receptor
- BBB
blood–brain barrier
- SD
standard deviation
Supporting Information Available
Experimental protocols and characterization data for the preparation of LSP1-2111, time course for brain and plasma exposures of LSP1-2111 using IP dosing, experimental parameters for the bioanalysis of LSP1-2111 in plasma, brain, ECF, and CSF, and efficacious doses of LSP1-2111 in all reported preclinical models. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Célanire S.; Campo B. Recent advances in the drug discovery of metabotropic glutamate receptor 4 (mGluR4) activators for the treatment of CNS and non-CNS disorders. Expert Opin. Drug Discovery 2012, 7, 261–280. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Hopkins C. R. Metabotropic glutamate receptor 4 (mGlu4)-positive allosteric modulators for the treatment of Parkinson’s disease: historical perspective and review of the patent literature. Expert Opin Ther. Pat. 2012, 22, 461–481. [DOI] [PubMed] [Google Scholar]
- Beurrier C.; Lopez S.; Révy D.; Selvam C.; Goudet C.; Lhérondel M.; Gubellini P.; Kerkerian-LeGoff L.; Acher F.; Pin J. P.; Amalric M. Electrophysiological and behavioral evidence that modulation of metabotropic glutamate receptor 4 with a new agonist reverses experimental parkinsonism. FASEB J. 2009, 23, 3619–3628. [DOI] [PubMed] [Google Scholar]
- Acher F.; Selvam C.; Pin J.-P.. Diastereoisomers of Hypophosphorous acid derivatives. World patent application WO 2010/106526. March 19, 2010.
- Flor P. J.; Acher F. C. Orthosteric versus allosteric GPCR activation: the great challenge of group-III mGluRs. Biochem. Pharmacol. 2012, 84, 414–424. [DOI] [PubMed] [Google Scholar]
- Goudet A. C.; Vilar B.; Courtiol T.; Deltheil T.; Bessiron T.; Brabet I.; Oueslati N.; Rigault D.; Bertrand H.-O.; McLean H.; Daniel H.; Amalric M.; Acher F.; Pin J.-P. A novel selective metabotropic glutamate receptor 4 agonist reveals new possibilities for developing subtype selective ligands with therapeutic potential. FASEB J. 2012, 26, 1682–1693. [DOI] [PubMed] [Google Scholar]
- Bennouar K. E.; Uberti M. A.; Melon C.; Bacolod M. D.; Jimenez H. N.; Cajina M.; Kerkerian-Le Goff L.; Doller D.; Gubellini P. Synergy between L-DOPA and a novel positive allosteric modulator of metabotropic glutamate receptor 4: implications for Parkinson’s disease treatment and dyskinesia. Neuropharmacology 2013, 66, 158–169. [DOI] [PubMed] [Google Scholar]
- Lopez S.; Bonito-Oliva A.; Pallottino S.; Acher F.; Fisone G. Activation of metabotropic glutamate 4 receptors decreases L-DOPA-induced dyskinesia in a mouse model of Parkinson’s disease. J. Parkinson’s Dis. 2011, 1, 339–346. [DOI] [PubMed] [Google Scholar]
- Wierońska J. M.; Stachowicz K.; Pałucha-Poniewiera A.; Acher F.; Brański P.; Pilc A. Metabotropic glutamate receptor 4 novel agonist LSP1-2111 with anxiolytic, but not antidepressant-like activity, mediated by serotonergic and GABAergic systems. Neuropharmacology 2010, 59, 627–634. [DOI] [PubMed] [Google Scholar]
- Wierońska J. M.; Stachowicz K.; Acher F.; Lech T.; Pilc A. Opposing efficacy of group III mGlu receptor activators, LSP1-2111 and AMN082, in animal models of positive symptoms of schizophrenia. Psychopharmacology 2012, 220, 481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierońska J. M.; Acher F. C.; Sławińska A.; Gruca P.; Łasoń-Tyburkiewicz M.; Papp M.; Pilc A. The antipsychotic-like effects of the mGlu group III orthosteric agonist, LSP1-2111, involves 5-HT1A signalling. Psychopharmacology 2013, 227, 711–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. J.; Iancu O. D.; Acher F. C.; Stewart B. M.; Eiwaz M A.; Duvoisin R. M.; Raber J. Role of mGluR4 in acquisition of fear learning and memory. Neuropharmacology 2013, 66, 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green A. R.; Gabrielsson J.; Fone K. C. F. Translational neuropharmacology and the appropriate and effective use of animal models. Br. J. Pharmacol. 2011, 164, 1041–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielsson J.; Green A. R.; Van der Graaf P. H. Optimising in vivo pharmacology studies—practical PKPD considerations. J. Pharmacol. Toxicol. Methods 2010, 61, 146–156. [DOI] [PubMed] [Google Scholar]
- Di L.; Rong H.; Feng B. Demystifying brain penetration in central nervous system drug discovery. J. Med. Chem. 2013, 56, 2–12. [DOI] [PubMed] [Google Scholar]
- Smith D.; Di L.; Kerns E. H. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discovery 2010, 9, 929–939. [DOI] [PubMed] [Google Scholar]
- In silico parameters calculated using MOE (Molecular Operating system, 2012).
- Wager T. T.; Chandrasekaran R. Y.; Hou X.; Troutman M. D.; Verhoest P. R.; Villalobos A.; Will Y. Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci. 2010, 1, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson M.; Løye A. F.; Mow T.; Hornberg J. J. A high content screening assay to predict human drug-induced liver injury during drug discovery. J. Pharmacol. Toxicol. Methods 2013, 68, 302–313. [DOI] [PubMed] [Google Scholar]
- Broad binding counterscreen conducted at Cerep (www.cerep.com).
- Broad functional counterscreens conducted at Euroscreen (www.euroscreen.com).
- Cremers T. I.; de Vries M. G.; Huinink K. D.; van Loon J. P.; v d Hart M.; Ebert B.; Westerink B. H.; De Lange E. C. Quantitative microdialysis using modified ultraslow microdialysis: direct rapid and reliable determination of free brain concentrations with the MetaQuant technique. J. Neurosci. Methods 2009, 178, 249–254. [DOI] [PubMed] [Google Scholar]
- For experimental conditions and bioanalytical methods for LSP1-2111 see Supporting Information.
- Rorick-Kehn L. M.; Perkins E. J.; Knitowski K. M.; Hart J. C.; Johnson B. G.; Schoepp D. D.; McKinzie D. L. Improved bioavailability of the mGlu2/3 receptor agonist LY354740 using a prodrug strategy: in vivo pharmacology of LY544344. J. Pharmacol. Exp Ther. 2006, 316, 905–13. [DOI] [PubMed] [Google Scholar]
- Triplett J. W.; Hayden T. L.; McWhorter L. K.; Gautam S. R.; Kim E.; Bourne E. D. W. A. Determination of gallium concentration in “blood-free” tissues using a radiolabeled blood marker. J. Pharm. Sci. 1985, 74, 1007–1009. [DOI] [PubMed] [Google Scholar]
- Monn J. A.; Massey S. M.; Valli M. J.; Henry S. S.; Stephenson G. A.; Bures M.; Hérin M.; Catlow J.; Giera D.; Wright R. A.; Johnson B. G.; Andis S. L.; Kingston A.; Schoepp D. D. Synthesis and metabotropic glutamate receptor activity of S-oxidized variants of (−)-4-amino-2-thiabicyclo-[3.1.0]hexane-4,6-dicarboxylate: Identification of potent, selective, and orally bioavailable agonists for mGlu2/3 receptors. J. Med. Chem. 2007, 50, 233–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.