

Abstract
A series of dibasic des-hydroxy β2 receptor agonists has been prepared and evaluated for potential as inhaled ultralong acting bronchodilators. Determination of activities at the human β-adrenoreceptors demonstrated a series of highly potent and selective β2 receptor agonists that were progressed to further study in a guinea pig histamine-induced bronchoconstriction model. Following further assessment by onset studies in guinea pig tracheal rings and human bronchial rings contracted with methacholine (guinea pigs) or carbachol (humans), duration of action studies in guinea pigs after intratracheal (i.t.) administration and further selectivity and safety profiling AZD3199 was shown to have an excellent over all profile and was progressed into clinical evaluation as a new ultralong acting inhaled β2 receptor agonist with rapid onset of action.
Keywords: uLABA, β2 receptor agonist, AZD3199, adrenoreceptor agonist, asthma, COPD, inhaled
The β2-adrenoceptor (β2AR) is a member of the class A, G protein-coupled receptor (GPCR) family and is widely distributed in the respiratory tract and particularly in airway smooth muscle. Agonists of this receptor are very effective bronchodilators,1 and in combination with either an inhaled corticosteroid or a muscarinic antagonist, β2AR agonists form the current standard for treatment for both asthma2 and chronic obstructive pulmonary disease COPD.3 The first generation β2AR agonists, e.g., salbutamol were classed as short duration β2AR agonists requiring multiple daily dosing. These were followed by second generation compounds (e.g., salmeterol or formoterol) that exhibited longer duration of action amenable to twice-daily dosing. Although these second generation inhaled β2AR agonists have proven very effective, poor compliance combined with ineffective control of nocturnal asthma have been recognized as issues limiting their clinical effectiveness. To address these concerns, third generation ultralong acting inhaled β2AR agonists, designed to have once-a-day duration of action, have been described4 including olodaterol5 and the launched products vilanterol6 and indacaterol7−8b (Figure 1).
Figure 1.

Selection of known β2AR agonists and sibenadet (a dual D2 dopamine receptor, β2-AR agonist).
The inhaled route to deliver β2AR agonists directly to the lung is used in order to both maximize the bronchodilator activity while minimizing systemic exposure of the compounds.9 In the clinical setting, the ability to deliver efficacy combined with an acceptable separation from the β2AR systemic-mediated side-effects is paramount in defining the predicted human dose. Therefore, a fine balance is required, where the drug candidate combines long retention in the target organ, thus enabling a long duration of action at the β2AR, combined with rapid clearance from the systemic circulation through the rapid elimination of either parent compound or associated metabolites after systemic redistribution of the compound from the lung tissues.
There has been much debate about the various proposed strategies for the rationalization of agents that exhibit a sustained duration of action when applied topically to the lung, and these hypotheses have been used to explain the observed differences in the duration of action seen in preclinical models. Examples of these rationalizations include among others:
(i) The diffusion microkinetic theory: where high membrane partitioning of lipophilic bases into membrane phospholipids is used to explain the long duration of action.10
(ii) The exosite binding theory: where it is proposed that a portion of the β2AR agonist (e.g., the lipophilic 4-phenylbutoxyhexyl group of salmeterol) interacts at a remote binding site in the β2-adrenoceptor away from the catechol binding site, and in doing so, holds the compound in close proximity to the receptor.11
(iii) High agonist intrinsic activity: resulting from high receptor occupancy giving prolonged pharmacological effect.12
(iv) Receptor kinetics: where slow receptor off-rates have been proposed to lead to enhanced duration of action in both inhaled β2-agonists and inhaled muscarinic M3 receptor antagonists.13
(v) Reduction in solubility and permeability: where slow dissolution into the airway smooth muscle affords the potential for extended lung retention.14
In addition to the aforementioned concepts, scientists from AstraZeneca recently published key concepts for extending the duration of action through optimization of the pharmacokinetics of the inhaled therapy.15
Coupled in part with the above hypotheses, increases in lipophilicity16 have been shown to be an important parameter in delivering compounds with an extended duration of action. Along with modulation in lipophilicity, it has been shown that the incorporation of a dibasic pharmacophore within the compounds leads to an increase in the duration of action of the β2AR agonists. However, it has also been suggested that, as you increase lipophilicity, the onset of action may be delayed.
A medicinal chemistry hypothesis was put in place to design and test a series of dibasic compounds, with a range of lipophilicities in order to determine if lipophilic dibasic compounds could have a short onset of action while retaining a once-a-day profile.
In addition to optimizing both the duration of action and fast onset, we monitored potential systemic β2AR agonist side-effects through β2AR-induced hypokalemia, an effect that is caused by increasing cellular uptake of potassium secondary to β2AR stimulation of sodium/potassium ATPase. The sodium/potassium ATPase is found predominantly on skeletal muscle and has a strong association with tremor. As a consequence, we decided to pursue plasma potassium levels as a marker of systemic β2AR agonist side-effects. At the outset of the project, we set the goal to identify an inhaled β2AR agonist that was well-tolerated, combined a rapid onset of action, comparable to formoterol to achieve better patient compliance,17 and would provide bronchoprotection from once-a-day dosing.
We wish to report the discovery of AZD3199, a new β2AR agonist that was designed to combine these project requirements.18 Recently we communicated the discovery of the new series of β2AR agonists that was devoid of the C-1 hydroxy group that is present in most β2AR agonists,19 and it has been shown both by ourselves and later by others that incorporation of a second basic group into the β2AR agonist template leads to compounds that demonstrate longer duration of action due in part to an increase in membrane partitioning.20,21 Design hypotheses were formulated to substitute the amide group (R2) with a range of aminoalkyl groups with the aim of generating a new series of potent β2AR agonists that possessed a long duration of action. Gratifyingly, the strategy delivered a range of dibasic compounds that maintained both the high potency and efficacy at the β2AR (compare potency of compounds 1 and 2 in Table 1). Exploration of the group R2 showed that a range of basic groups were accommodated without detrimental effect on potency (2–6 and 8–11); however, because of balancing the overall properties, compounds containing the N,N-diethylamino ethyl group such as for compound 2 was chosen for further optimization. Within this series, limited exploration of the phenethyl group (R3) showed a surprising effect on efficacy (cf. examples 2 and 12–16), and eventually 12 (AZD3199) was progressed for further evaluation. Interestingly, the installation of the chiral C-1 hydroxy group had little effect on potency and agonism (cf. examples 2 and 3; 4 and 16), and these C-1 hydroxy containing compounds were not progressed further.
Table 1. Potency and Intrinsic Activity at the Human β2AR.
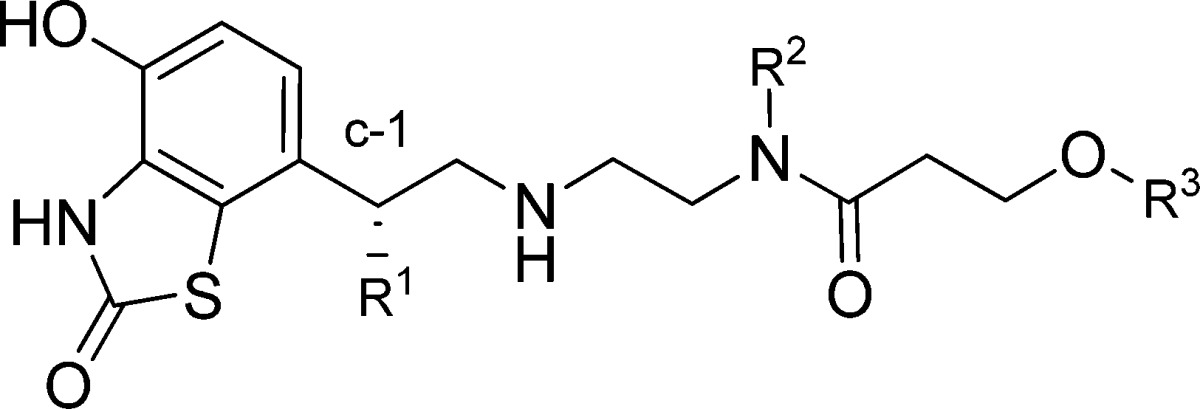
| compd no. | R1 | R2 | R3 | β2 potency pEC50a | IAb |
|---|---|---|---|---|---|
| 1 | H | nBu | phenethyl | 8.8 | 0.9 |
| 2 | H | -(CH2)2NEt2 | phenethyl | 8.1 | 0.9 |
| 3 | OH | -(CH2)2NEt2 | phenethyl | 8.4 | 1.0 |
| 4 | OH | -(CH2)2NEt2 | 3-chlorophenethyl | 8.4 | 0.9 |
| 5 | H | -(CH2)2NMe2 | phenethyl | 8.2 | 1.0 |
| 6 | H | -(CH2)3NH2 | phenethyl | 6.5 | 0.5 |
| 7 | H | -(CH2)3NMe2 | phenethyl | 7.8 | 1.0 |
| 8 | H | -(CH2)3N(Me)iPr | 3-chlorophenethyl | 8.3 | 0.8 |
| 9 | H | -(CH2)3N(Me)Et | 3-chlorophenethyl | 8.0 | 0.8 |
| 10 | H | -(CH2)3N(Me)cyPr | 3-chlorophenethyl | 8.1 | 0.8 |
| 11 | H | -(CH2)2-piperidin-1-yl | 3-chlorophenethyl | 7.9 | 0.7 |
| 12 | H | -(CH2)2NEt2 | 2-(naphthalen-1-yl)ethyl | 8.2 | 0.8 |
| 13 | H | -(CH2)2NEt2 | 2-(naphthalen-2-yl)ethyl | 8.0 | 0.5 |
| 14 | H | -(CH2)2NEt2 | 3-fluorophenethyl | 8.1 | 0.8 |
| 15 | H | -(CH2)2NEt2 | 3-methoxyphenethyl | 8.1 | 0.6 |
| 16 | H | -(CH2)2NEt2 | 3-chlorophenethyl | 8.2 | 0.7 |
β2AR agonism was performed in H292 cells (bronchial epithelial cell line) expressing the human β2 adrenergic receptor. Functional activity was determined by measuring accumulation of intracellular cAMP using AlphaScreen. The compounds were incubated for 1 h at 22 °C.
IA (Intrinsic activity) measured relative to formoterol (pEC50 8.6, IA = 1).
Pharmacological selectivity margins were calculated relative to the human β2 cAMP potency determined in H292 cells. Surprisingly, the incorporation of the basic group (R2) gave compounds with an excellent β2-AR selectivity, and AZD3199 has greater than 1000-fold binding selectivity over β1 and β3 ARs (Chart 1).
Chart 1. Binding selectivity for AZD3199 vs. β1 and β3a.
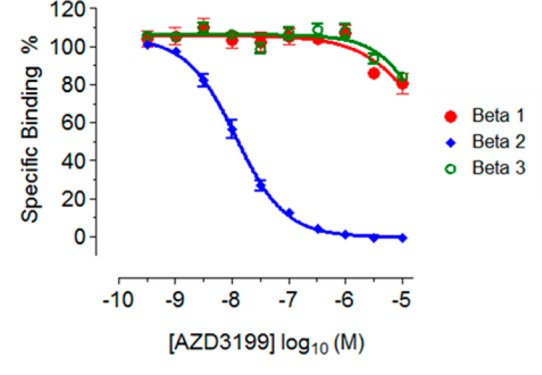
a Radioligand competition binding assays were determined for [125I]-iodocyanopindolol binding to membranes expressing beta adrenergic receptors. Compound inhibition was determined following incubation for 2 h at 22 °C and expressed as percentage inhibition relative to the control.
Further selectivity profiling against alpha adrenoreceptors (α1D) and D2 dopamine receptors (D2s) demonstrated high selectivity for AZD3199 (Table S1 Supporting Information). AZD3199 was greater than 100-fold selective over the D2s receptor and 50-fold selective against the α1D receptor and showed no agonism at this receptor even though the recombinant calcium assay employed was considered to be a high receptor reserve system relative to human vascular tissue. At the D2s receptor, AZD3199 had 32-fold selectivity and was a partial agonist with respect to sibenadet (a dual D2 dopamine receptor, β2-AR agonist). The agonist potency of AZD3199 at D2s was 100-fold less than sibenedet (pEC50 = 8.7). In contrast to the other compounds (see Table S1 Supporting Information), which were all full agonists at the β1AR, AZD3199 showed no agonism at the β1AR even though the β1 recombinant assay used in this study was considered to be a high receptor reserve system when compared with human in vitro tissue experiments in the literature.
AZD3199 was shown to have high potency and intrinsic activity for the β2AR when assessed for different species across sources and assay formats (see Table S2 Supporting Information). The functional activity at the guinea pig β2AR was determined for AZD3199 and other reference compounds by measuring relaxation of tracheal rings preconstricted with methacholine in an organ bath. The potency was measured as the EC50 concentration (Table 2).
Table 2. Potency β2-Adrenoceptor Agonism in Isolated Guinea Pigs Tissue.
| compd | guinea pigs pA50 | guinea pigs intrinsic activity |
|---|---|---|
| AZD3199 | 8.0 | 0.8 |
| indacaterol | 7.7 | 0.7 |
| formoterol | 8.8 | 0.9 |
| salmeterol | 7.7 | 0.5 |
| salbutamol | 6.7 | 0.6 |
Onset of relaxation was measured in vitro using guinea pig tracheal rings and human bronchial rings contracted with methacholine (guinea pigs) or carbachol (humans).22 The EC50 concentration was given and the relaxation followed over time. The time taken to reach 90% of the final relaxation was defined as the onset time (Chart 2).
Chart 2. Onset of Relaxation of AZD3199 As Measured Using Guinea Pig Tracheal Rings Contracted with 1 μM Methacholine.
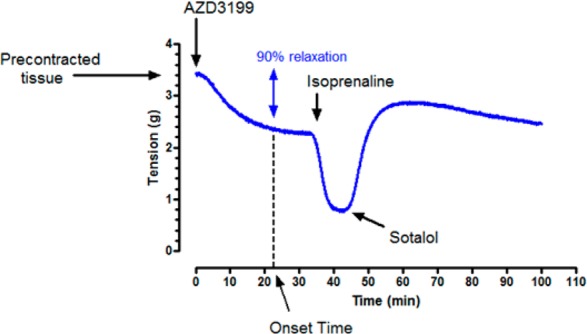
A single concentration of AZD3199 was given (at or about the pEC50 concentration) and the relaxation was followed over time. After AZD3199 had reached a plateau, isoprenaline was added to confirm that the concentration of AZD3199 used was at EC50 for the tissue. Finally, the addition of 10 μM sotalol was used to demonstrate reversibility of the response.
AZD3199 was shown to have a fast onset time, comparable to formoterol, in both guinea pigs (22 min) and humans (11 min), significantly faster than that of salmeterol (Table 3).
Table 3. Potency and Onset Time of β2-Adrenoceptor Agonism in Isolated Tissuea.
| compd | guinea pig onset time (min) | human onset time (min) | cLogP | type |
|---|---|---|---|---|
| AZD3199 | 22 | 11 | 6.0 | dibasic |
| 1 | 90 | n/d | 4.5 | monobasic |
| 7 | 6 | n/d | 3.7 | dibasic |
| 14 | 13 | n/d | 4.9 | dibasic |
| 16 | 11 | n/d | 5.5 | dibasic |
| sibenadet | 58 | n/d | 2.5 | monobasic |
| indacaterol | 77 | 49 | 3.0 | monobasic |
| formoterol | 23 | 13 | 1.3 | monobasic |
| salmeterol | 90 | t/s | 3.1 | monobasic |
| salbutamol | 3 | 19 | 0.1 | monobasic |
n/d = not determined; t/s = onset time too slow to measure.
AZD3199 and other selected reference compounds were progressed to the guinea pig histamine-induced bronchoconstriction model, a well characterized species for modeling human lung disease.23 AZD3199 was given, and a clear dose–response curve was seen (Chart S1 Supporting Information); the ED80 dose was determined as 27 μg/kg (46.7 nmol/kg). Propanolol (1 mg/kg i.v.) reversed this bronchoprotection confirming that the activity was mediated via β2 receptors.
A plot of lipophilicity (clogP) as a function of guinea pig onset time showed a very interesting correlation and demonstrated that for monobasic compounds a strong correlation exists between increasing onset time with increasing lipophilicity. However, data suggests that for dibasic compounds, a much reduced onset time could be achieved with compounds that have increased lipophilicity (Chart 3).
Chart 3. Plot of Guinea Pig Onset Time vs. cLogP for a Selection of Monobasic or Dibasic Compounds.

On the basis of encouraging in vitro profiles [rat intrinsic clearance (105 μL/min/million cells)] as well as good level of potency and intrinsic activity in guinea pig tissues, AZD3199 was progressed to in vivo rat intravenous (i.v.) pharmacokinetic (PK) profiling in order to determine the terminal half-life. Indeed, in the course of this project it was shown that in vivo rat plasma i.v. t1/2 can be used as a predictor of in vivo duration in the bronchoprotection guinea pig model following intratracheal (i.t.) dosing (cf. plasma t1/2 > 10 h for 24 h pharmacokinetic duration).24,25 AZD3199 has i.v. plasma terminal half-lives of 11, 17, and 18 h in rats, guinea pigs, and dogs, respectively, and combined with high volumes of distribution (Vz) 23 (rats), 22 (guinea pigs), and 17 (dogs) L/kg afforded confidence of seeing a long duration of action. Therefore, on the basis of its overall profile, AZD3199 was selected for further progression to duration studies in a guinea pig bronchoprotection model. The lung terminal half-life was predicted from the rat i.v. dosing giving confidence of potential for u.i.d. dosing. The low oral availability of AZD3199 in both rats and dogs (F < 2%) would limit any systemic exposure due to swallowed fraction.
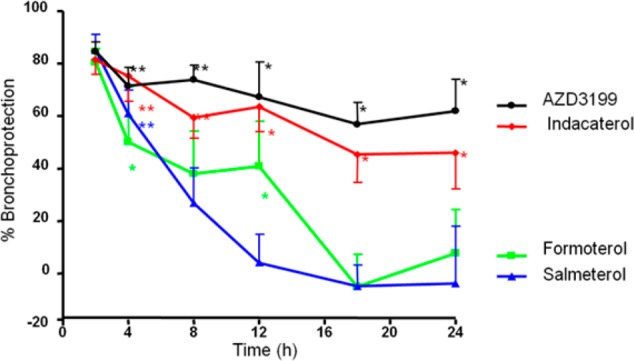
The durations of action of AZD3199 and assorted reference compounds were measured in guinea pigs by the i.t. route. Guinea pigs were given the ED80 dose of compound, and at various time points after dosing, the inhibition of histamine-induced bronchoconstriction were measured. AZD3199 retained 58% bronchoprotection 24 h after i.t. dosing, significantly different to control animals. The results for AZD3199, indacaterol, formoterol, and salmeterol are shown (Chart 4). AZD3199 clearly had a longer duration of action than formoterol and salmeterol and a similar duration of action to indacaterol.
Chart 4. Duration of Action Studies in Guinea Pigs for AZD3199 and a Selection of Standard Compounds.
β2AR agonists have several mechanistic side effects due to activation of peripheral β2 receptors: tachycardia, QTc changes, hypertension, hypokalaemia, tremor, and hyperglycaemia; and several of these were measured after administration of salbutamol. From these experiments, plasma potassium was chosen as the marker of choice. The effects were investigated using infusions of salbutamol, formoterol, and AZD3199 in anaesthetized guinea pigs. The infusions were designed to give a constant plasma level between 30 and 60 min after the start of the infusion. Dose-related changes in plasma potassium and other markers were seen as expected. Potassium levels were compared at 60 min and plotted against the plasma level of compound, corrected for plasma protein binding and efficacy (see Chart S2 Supporting Information). At the lowest plasma level, AZD3199 produced a significantly smaller reduction in plasma potassium compared to formoterol; at all other plasma levels there were no differences between the compounds. These data suggest that in guinea pigs the mechanistic side effects of AZD3199 are no worse than formoterol and are significantly better at low plasma levels.
AZD3199 was prepared according to the method outlined in Scheme 1. The commercial compound 17 was reacted with the acid chloride prepared in situ from acid 18(26) to afford alcohol 19. A 2-step oxidation followed by a reductive amination procedure with the known amine 20(27) afforded 12 (AZD3199) in reasonable over all yield.
Scheme 1. Chemical synthesis of AZD3199.

Reagents and conditions: (a) 18 + (COCl)2 (1.1 mol equiv), dimethyl formamide (cat.), CH2Cl2, rt, 15 h, concentrate and add to 17, Hunig’s base (2 mol equiv), CH2Cl2, rt, 20 h (75%). (b) (i) dimethyl sulfoxide (2.2 mol equiv), CH2Cl2, −78 °C, add (COCl)2 (1.1 mol equiv), stir 15 min; (ii) add 19, stir 15 min; (iii) add triethylamine (5 mol equiv) and warm to rt 90 min; (iv) 20 in CH2Cl2, sodium triacetoxyborohydride (2 mol equiv), rt (27%).
In summary, a new series of dibasic C-1 des-hydroxy 7-hydroxy benzthiazolone β2AR agonists have been designed to combine high potency, long duration of action and fast onset of action. From the design series, AZD3199 was shown to be highly selective (>1500-fold) for the human β2AR (pEC50 7.9 ± 0.12 (n = 8)) over human β1- and β3-ARs. Further profiling demonstrated AZD3199 to be highly potent when dosed in vivo in a guinea pig histamine-induced bronchoconstriction model, exhibiting long duration of action amenable to a once-a-day dosing regimen. We have demonstrated that a short onset time can be achieved within the series through combining high lipophilicity and two basic groups. Utilizing plasma potassium levels in a guinea pig model as a marker of potential β2AR-induced systemic side effects, AZD3199 produced, at the lowest plasma level, a significantly smaller reduction in plasma potassium compared to formoterol; at all other plasma levels there were no differences between the compounds. These data suggest that in guinea pigs the mechanistic side effects of AZD3199 are no worse than formoterol and are significantly better at low plasma levels. In conclusion, AZD3199 is a new ultralong acting inhaled β2AR agonist and further work will be disclosed shortly.
Acknowledgments
We thank Dr. Richard Lewis, John Steele, Mike Bernstein, and Peter Sjö for assistance in submitting the communication.
Glossary
ABBREVIATIONS
- β2AR
β2-adrenoceptor
- GPCR
G Protein-coupled receptor
- COPD
chronic obstructive pulmonary disease
Supporting Information Available
Synthetic procedures, analytical data, tables S1, S2, and S3, and Charts S1 and S2 including procedures for pharmacological activities. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ (M.J.S.) Centre for Biomolecular Sciences, School of Pharmacy, The University of Nottingham, University Park, Nottingham NG7 2RD, U.K.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Fuso L.; Mores N.; Valente S.; Malerba M.; Montuschi P. Long-acting beta-agonists and their association with inhaled corticosteroids in COPD. Curr. Med. Chem. 2013, 20, 1477–1495. [DOI] [PubMed] [Google Scholar]
- Walker J. K. L.; Penn R. B.; Hanania N. A.; Dickey B. F.; Bond R. A. New perspectives regarding β(2)-adrenoceptor ligands in the treatment of asthma. Br. J. Pharmacol. 2011, 163, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola M.; Page C. P.; Rogliani P.; Matera M. G. β2-agonist therapy in lung disease. Am. J. Respir. Crit. Care Med. 2013, 187, 690–696. [DOI] [PubMed] [Google Scholar]
- Matera M. G.; Cazzola M. Ultra-long-acting beta2-adrenoceptor agonists: an emerging therapeutic option for asthma and COPD?. Drugs 2007, 67, 503–515. [DOI] [PubMed] [Google Scholar]
- Bouyssou T.; Hoenke C.; Rudolf K.; Lustenberger P.; Pestel S.; Sieger P.; Lotz R.; Heine C.; Buettner F. H.; Schnapp A.; Konetzki I. Discovery of olodaterol, a novel inhaled β2-adrenoceptor agonist with a 24 h bronchodilatory efficacy. Bioorg. Med. Chem. Lett. 2010, 20, 1410–1414. [DOI] [PubMed] [Google Scholar]
- Procopiou P. A.; Barrett V. J.; Bevan N. J.; Biggadike K.; Box P. C.; Butchers P. R.; Coe D. M.; Conroy R.; Emmons A.; Ford A. J.; Holmes D. S.; Horsley H.; Kerr F.; Li-Kwai-Cheung A.-M.; Looker B. E.; Mann I. S.; McLay I. M.; Morrison V. S.; Mutch P. J.; Smith C. E.; Tomlin P. Synthesis and structure–activity relationships of long-acting β2 adrenergic receptor agonists incorporating metabolic inactivation: an antedrug approach. J. Med. Chem. 2010, 53, 4522–4530. [DOI] [PubMed] [Google Scholar]
- Davies S. L.; Castaner J. Indacaterol. Drugs Future 2005, 30, 1219–1224. [Google Scholar]
- Roig J.; Hernando R.; Mora R. Indacaterol, a novel once daily inhaled β2-adrenoreceptor agonist.. Open Respir. Med. J. 2009, 3, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur F.; Beattie D.; Beer D.; Bentley D.; Bradley M.; Bruce I.; Charlton S. J.; Cuenoud B.; Ernst R.; Fairhurst R. A.; Faller B.; Farr D.; Keller T.; Fozard J. R.; Fullerton J.; Garman S.; Hatto J.; Hayden C.; He H.; Howes C.; Janus D.; Jiang Z.; Lewis C.; Loeuillet-Ritzler F.; Moser H.; Reilly J.; Steward A.; Sykes D.; Tedaldi L.; Trifilieff A.; Tweed M.; Watson S.; Wissler E.; Wyss D. The identification of indacaterol as an ultralong-acting inhaled β2-adrenoceptor agonist. J. Med. Chem. 2010, 53, 3675–3684. [DOI] [PubMed] [Google Scholar]
- Mortimer K. J.; Harrison T. W.; Tang Y.; Wu K.; Lewis S.; Sahasranaman S.; Hochhaus G.; Tattersfield A. E. Plasma concentrations of inhaled corticosteroids in relation to airflow obstruction in asthma. Br. J. Clin. Pharmacol. 2006, 62, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G. P.; Lindén A.; Rabe K. F. Why are long-acting beta-adrenoceptor agonists long-acting?. Eur. Respir. J. 1994, 7, 569–578. [DOI] [PubMed] [Google Scholar]
- Green S. A; Spasoff A. P.; Coleman R. A.; Johnson M.; Liggett S. B. Sustained activation of a G protein-coupled receptor via ″anchored″ agonist binding. Molecular localization of the salmeterol exosite within the β2-adrenergic receptor. J. Biol. Chem. 1996, 39, 24029–24035. [DOI] [PubMed] [Google Scholar]
- Bao Y.; Horst L. Action mechanism of long- acting bronchodilators. Acta Pharm. Sin. 1998, 20, 191–196. [PubMed] [Google Scholar]
- Deyrup M. D.; Nowicki S. T.; Richards N. G. J.; Otero D. H.; Harrsison J. K.; Baker S. P. Structure-affinity profile of 8-hydroxycarbostyril-based agonists that dissociate slowly from the beta2-adrenoceptor. Arch. Pharmacol. 1999, 359, 168–177. [DOI] [PubMed] [Google Scholar]
- Patton J. S.; Byron P. R. Inhaling medicines: delivering drugs to the body through the lungs. Nat. Rev. Drug Discovery 2007, 6, 67–74. [DOI] [PubMed] [Google Scholar]
- Cooper A. E.; Ferguson D.; Grime K. Optimization of DMPK by the inhaled route: challenges and approaches. Curr. Drug. Metab. 2012, 13, 457–473. [DOI] [PubMed] [Google Scholar]
- Alikhani V.; Beer D.; Bentley D.; Bruce I.; Cuenoud B. M.; Fairhurst R. A.; Gedeck P.; Haberthuer S.; Hayden C.; Janus D.; Jordan L.; Lewis C.; Smithies K.; Wissler E. Long-chain formoterol analogues: an investigation into the effect of increasing amino-substituent chain length on the β2-adrenoceptor activity. Bioorg. Med. Chem. Lett. 2004, 14, 4705–4710. [DOI] [PubMed] [Google Scholar]
- White M. V.; Sander N. Asthma from the perspective of the patient. J. Allergy Clin. Immunol. 1999, 104, s47–52. [DOI] [PubMed] [Google Scholar]
- Kuna P.; Ivanov Y.; Trofimov V. I.; Saito T.; Beckman O.; Bengtsson T.; Jorup C.; Maltais F. Efficacy and safety of AZD3199 vs formoterol in COPD: a randomized, double-blind study. Respir. Res. 2013, 14, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocks M. J.; Alcaraz L.; Bailey A.; Bonnert R.; Cadogan E.; Christie J.; Connolly S.; Cook A.; Fisher A.; Flaherty A.; Hill S.; Humphries A.; Ingall A.; Jordan S.; Lawson M.; Mullen A.; Nicholls D.; Paine S.; Pairaudeau G.; St-Gallay S.; Young A. Design driven HtL: The discovery and synthesis of new high efficacy β2-agonists. Bioorg. Med. Chem. Lett. 2011, 21, 4027–4031. [DOI] [PubMed] [Google Scholar]
- Austin R. P.; Barton P.; Bonnert R. V.; Brown R. C.; Cage P. A.; Cheshire D. R.; Davis A. M.; Dougall I. G.; Ince F.; Pairaudeau G.; Young QSAR and the rational design of long-acting dual D2 -receptor/β2-adrenoceptor agonists. J. Med. Chem. 2003, 46, 3210–3220. [DOI] [PubMed] [Google Scholar]
- Alcaraz L.; Bailey A.; Cadogan E.; Connolly S.; Jewell R.; Jordan S.; Kindon N.; Lister A.; Lawson M.; Mullen A.; Dainty I.; Nicholls D.; Paine S.; Pairaudeau G.; Stocks M. J.; Thorne P.; Alan Young A. From libraries to candidate: the discovery of new ultra long-acting dibasic β2-adrenoceptor agonists. Bioorg. Med. Chem. Lett. 2012, 22, 689–695. [DOI] [PubMed] [Google Scholar]
- Procopiou P. A.; Barrett V. J.; Biggadike K.; Butchers P. R.; Craven A.; Ford A. J.; Guntrip S. B.; Holmes D. S.; Hughes S. C.; Jones A. E.; Looker B. E.; Mutch P. J.; Ruston M.; Needham D.; Smith C. E. Discovery of a rapidly metabolized, long-acting β2 adrenergic receptor agonist with a short onset time incorporating a sulfone group suitable for once-daily dosing. J. Med. Chem. 2014, 57, 159–170. [DOI] [PubMed] [Google Scholar]
- Walland A.; Palluk R.; Burkard S.; Hammer R. Compensation of muscarinic bronchial effects of talsaclidine by concomitant sympathetic activation in guinea pigs. Eur. J. Pharmacol. 1997, 330, 213–219. [DOI] [PubMed] [Google Scholar]
- Connolly S.; Alcaraz L.; Bailey A.; Cadogan E.; Christie J.; Cook A. R.; Fisher A. J.; Hill S.; Humphries A.; Ingall A. H.; Kane Z.; Paine S.; Pairaudeau G.; Stocks M. J.; Young A. Design-driven LO: the discovery of new ultra long acting dibasic β2-adrenoceptor agonists. Bioorg. Med. Chem. Lett. 2011, 21, 4612–4616. [DOI] [PubMed] [Google Scholar]
- Tayab Z. R.; Hochhaus G. Pharmacokinetic/pharmacodynamic evaluation of inhalation drugs: application to targeted pulmonary delivery systems. Expert Opin. Drug. Delivery 2005, 2, 519–532. [DOI] [PubMed] [Google Scholar]
- Bailey A.; Bonnert R.; Flaherty A.; Pairaudeau G.; Stocks M.. WO 2007027133, 2007.
- Giles M. E.; Thomson C.; Eyley S. C.; Cole A. J.; Goodwin C. J.; Hurved P. A.; Morlin A. J. G.; Tornos J.; Atkinson S.; Just C.; Dean J. C.; Singleton J. T.; Longton A. J.; Woodland I.; Teasdale A.; Gregertsen B.; Else H.; Athwal M. S.; Tatterton S.; Knott J. M.; Thompson N.; Smith S. Development of a manufacturing process for sibenadet hydrochloride, the active ingredient of Viozan. J. Org. Process Res. Dev. 2004, 8, 628–642. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.