

Abstract

The in silico construction of a PDGFRβ kinase homology model and ensuing medicinal chemistry guided by molecular modeling, led to the identification of potent, small molecule inhibitors of PDGFR. Subsequent exploration of structure–activity relationships (SAR) led to the incorporation of a constrained secondary amine to enhance selectivity. Further refinements led to the integration of a fluorine substituted piperidine, which resulted in significant reduction of P-glycoprotein (Pgp) mediated efflux and improved bioavailability. Compound 28 displayed oral exposure in rodents and had a pronounced effect in a pharmacokinetic–pharmacodynamic (PKPD) assay.
Keywords: Platelet-derived growth factor, fluoro-piperidine, fluorine, Pgp
Platelet-derived growth factor receptor (PDGFR) is an attractive target for treating malignant disease.1,2 Constitutive activation of PDGFR resulting from point mutations, chromosomal translocations, and autocrine loops have been described in gastrointestinal stromal tumors (GIST) and gliomas.3 PDGFR also plays an important role in the remodeling and maintenance of blood vessels (neovasculature) through its regulation of pericytes, suggesting that inhibitors of PDGFR could have broad utility against a spectrum of human cancers as antiangiogenic agents.4,5 In addition, PDGFR has been shown to be a potent mitogen for myofibroblast formation, a signature event in the progression of fibrosis.6 The availability of a potent, selective, orally bioavailable PDGFR inhibitor would help define the role of the kinase in tumor growth, angiongenesis and fibrosis.
Although a few approved kinase inhibitors possess PDGFR inhibitory activity, their primary activity is targeted toward Abl or KDR, and dose escalation is often limited by attendant toxicities (i.e., Imatinib, Dasatinib, Sorafenib, Sunitinib, etc.). Prior to our investigation, two selective small-molecule inhibitors of PDGFR had been described in the literature.7−9 We set out to discover and develop a novel, orally bioavailable, selective, and potent PDGFR antagonist using structure-based drug design strategies.
PDGFR belongs to the receptor tyrosine kinase class 3 (RTKIII), which comprises members of the PDGFR family, including PDGFRα, PDGFRβ, cKIT, cFMS, and FLT3. A highly related group of kinases include the VEGFR family, FLT1, FLT4, and KDR.10−12 Although the sequence of PDGFR is known, there are no reported PDGFR crystal structures. The Protein Data Bank (www.pdb.org) does, however, include crystal structures of other members of the PDGFR and VEGFR families, namely, cFMS, cKIT, FLT3, KDR (VEGFR2), and FLT1 (VEGFR1). Many of these crystal structures also contain small molecule inhibitors cocrystallized in the ATP binding site. From this information, we can discern that there are minor protein sequence variations within the ATP binding pockets of these kinase families.13 Our approach was to take advantage of the differences in sequence as well as the observed subtle differences in ATP-binding site shapes in order to impart the desired PDGFR selectivity to our inhibitors.
By using a published cFMS crystal structure and the known amino acid sequence of PDGFRβ, we constructed a model of PDGFRβ in silico. This model was then used to design novel small molecule antagonists containing an imidazopyridine to interact with the hinge region of PDGFRβ (Figure 1). Docking studies predicted that the addition of an amine would provide an interaction with the side chain of either an aspartic acid (i.e., ASP-111) or an asparagine (i.e., ASN-253) in the outer floor. The backbone carbonyl of arginine-252 was predicted to provide another polar interaction for a small molecule inhibitor. These interactions were predicted to provide compounds with good PDGFRβ potency, as well as potentially provide selectivity over members of the VEGFR family based on the expected variation in protein structure between kinases.13 This effort culminated in the design of a novel series of imidazopyridines.14
Figure 1.
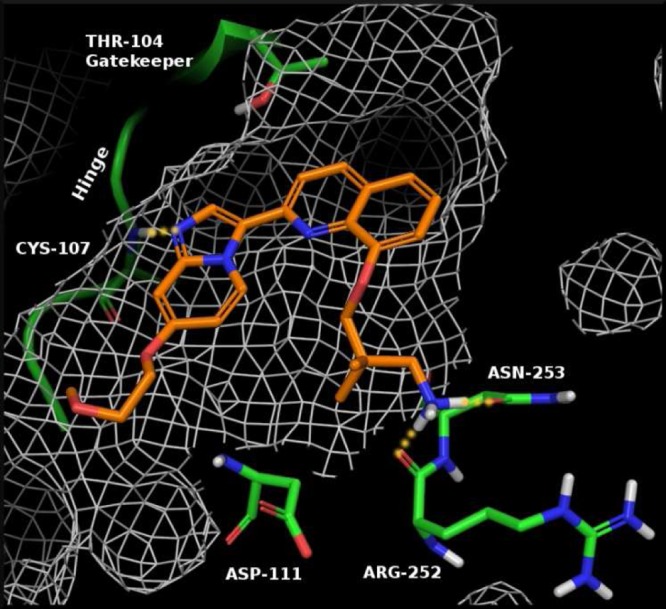
View of 1 docked into the ATP binding site of a PDGFRβ kinase homology model. Location of PDGFR residues are labeled for clarity.
An early example of this series (1, see Figures 1 and 2) demonstrated an IC50 of 18 nM in our PDGFRβ cell assay.15 A molecular modeling view of compound 1 bound to the active site of PDGFRβ is shown in Figure 1, with the key residues highlighted. Although compound 1 was a potent PDGFRβ antagonist, selectivity over other members of the PDGFRβ and VEGFR families was limited (FLT3, cFMS, and KDR = <10×). We hypothesized that this poor selectivity was due to the high degree of conformational flexibility of the propyl-amine, allowing for a variety of polar interactions to be established within familial kinases. Consequently, we designed constrained analogues of 1 that could procure improved kinase selectivity (>10×) based on PDGFRβ specific residues.
Figure 2.

Structure and activity profile for compound 1.
With the identification of 1, our medicinal chemistry efforts focused on the development of a synthetic route that would allow for late stage diversification of the amine headgroup. This effort resulted in a highly convergent synthesis that began with commercially available nitropyridine 2, which was treated with 2-methoxyethanol in the presence of a strong base to provide chloropyridine 3, which was used directly in the next step without purification. Compound 4 was obtained by the treatment of 3 with LiHMDS in the presence of Pd and X-Phos, followed by exposure to aqueous acid.16 Cyclization of 4 was performed with aqueous chloroacetaldehyde at 50 °C to afford the key imidazopyridine 5. The quinoline core was obtained by starting with commercially available quinoline diol 6, which was exposed to benzyl bromide in the presence of K2CO3 to provide 7. Treatment of quinolinol 7 with oxalyl chloride and catalytic DMF in DCE at 85 °C provided the coupling partner 8 (Scheme 1).
Scheme 1. Synthesis of Key Coupling Partners.

With the key partners in hand we were able to couple 5 and 8 via a Heck-type reaction using a mixed catalyst system of Pd(PPh3)4 and Pd(OAc)2 in dioxane and water.17,18 The residual benzyl protecting group was then removed using Pd(OH)2 on carbon in the presence of ammonium formate to afford 9 (Scheme 2). This useful, advanced intermediate was conveniently alkylated with mesylates such as 10 using Cs2CO3 in DMA to afford final products having an amine headgroup, after TFA-mediated removal of the BOC protecting group. This synthetic sequence was highly convergent and allowed for robust generation of diverse structure–activity relationships (SAR) around the amine headgroup, providing compounds such as 11.
Scheme 2. Coupling and Alkylation Endgame of Synthesis.

With the development of a robust synthesis for the formation of 9, we began our refinement of 1 by searching for a constrained amine headgroup that would provide the desired selectivity (>10×) over members of the PDGFRβ and VEGFR families (Table 1). In general, the spatial orientation of the amine played an important role toward PDGFRβ potency and selectivity. With the aid of our PDGFRβ homology model, we found that it was possible to design an amine headgroup that would provide good PDGFRβ potency and improved kinase selectivity when compared to 1. Surprisingly, some amine headgroups were found to provide analogues having excellent selectivity over cFMS and cKIT (>100×), despite the same amino acid residues being located at the outer floor of the binding pocket. This is potentially due to the specific location of the polar residues in the outer floor of PDGFRβ, such as ASP-111 and ARG-252, and our use of a rigid amine headgroup. In addition, we found that >10× selectivity15 over KDR could be obtained with a variety of amine headgroups (14, 15, 18, 21, and 22), with the observed increase in PDGFRβ potency likely due to optimized binding with the polar residues in the outer floor. Some flexibility in the distance between the quinolone and amine NH was tolerated, with 2 to 3 carbon atoms between the quinoline oxygen and amine appearing optimal in cyclized amines (i.e., 11 and 15) but an optimal length was observed (11 vs 19). Secondary cyclic amines were preferred over primary acyclic amines (1 and 26 vs 11). It was found that pyrrolidine headgroups were well tolerated (21 and 22) except when the carbon adjacent to the amine was substituted (23 and 24). Alkylation of the amine (12) decreased PDGFRβ potency while leaving KDR potency unchanged. In addition, it was determined that the absolute configuration of a chiral amine can play a significant role in potency (15 vs 16).
Table 1. In Vitro Potency of Compounds 11–26a.
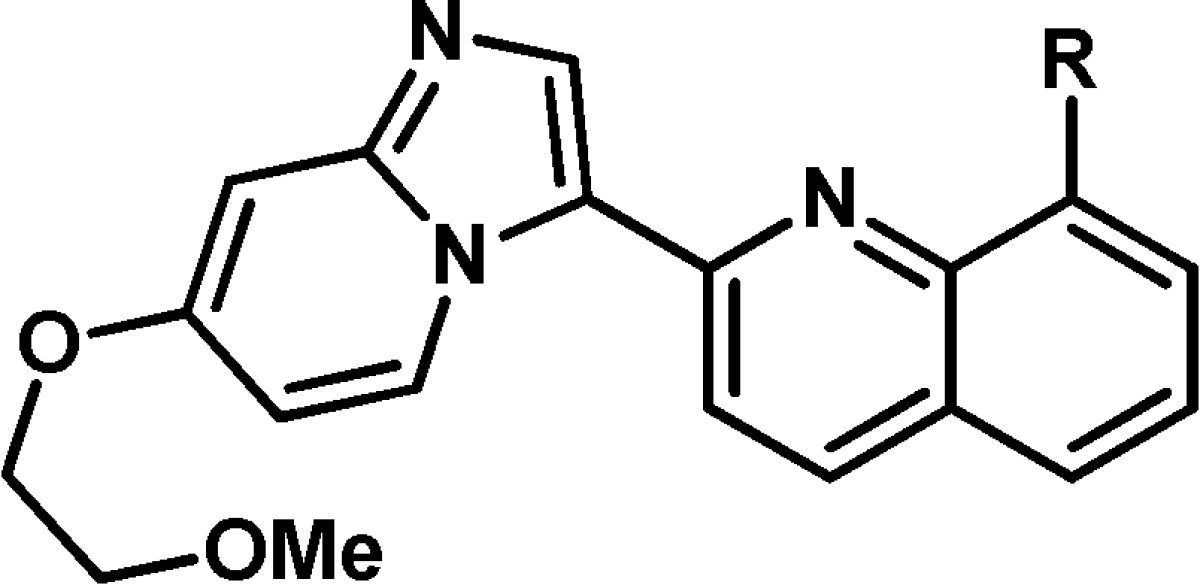
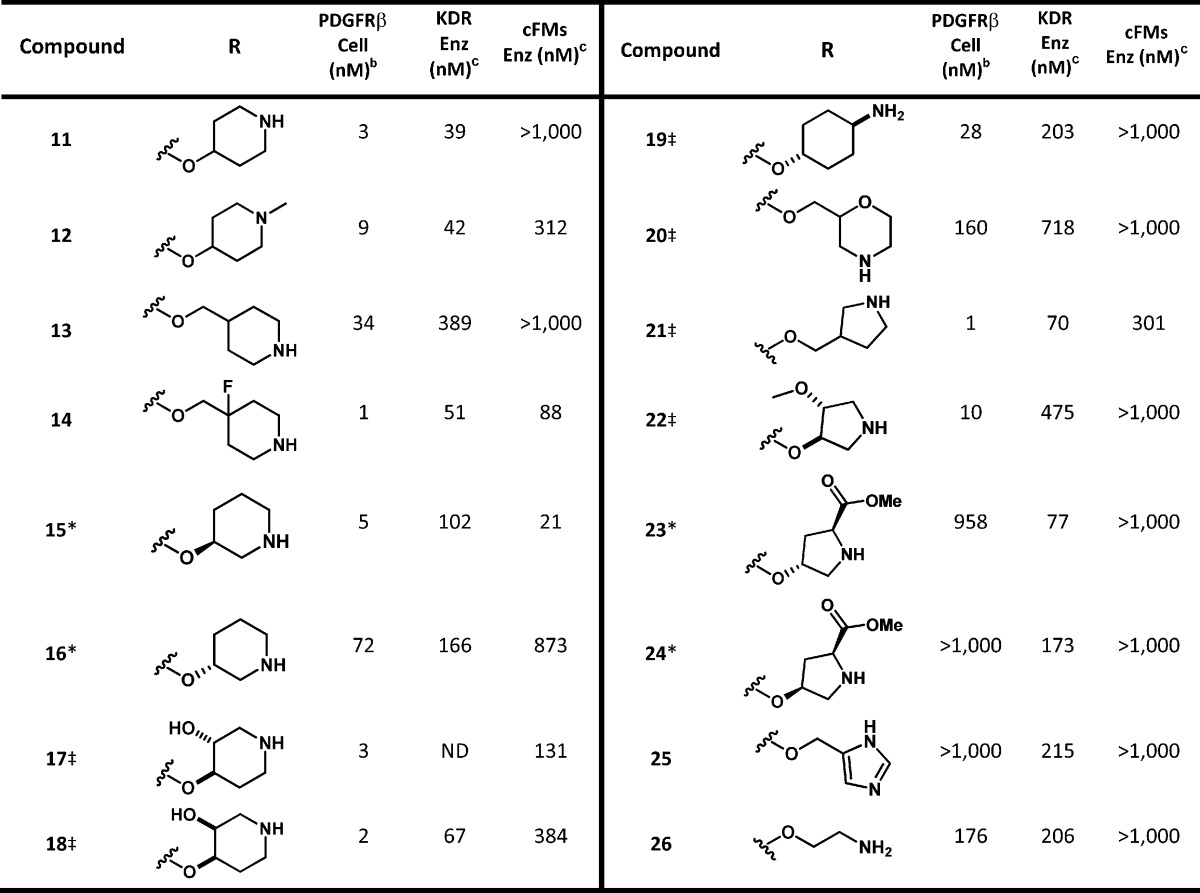
Key: ‡, denotes racemic compound, relative stereochemistry is as depicted; *, denotes single enantiomer. ND = not determined.
All compounds with PDGFR IC50 values of <100 nM were run at least in duplicate.
All compounds were run in this assay at least in duplicate; value shown is an average of the assay results.
After substantial effort we determined that compound 11 had achieved the target properties of potency and selectivity (>10× over KDR and >100× over cFMS, see Table 1) providing the desired improvements over 1.15 Although we had discovered alternative compounds that were more potent than 11, the overall in vitro properties of 11 were superior. This determination was based on a combination of properties, including stability (i.e., 14 had >70% microsomal clearance in rodents and cyno), low cell shift in the presence of human plasma (i.e., PDGFRβ cellular assay run in the presence of 50% human plasma; 21 = 29 nM, 22 = >1000 nM), and selectivity over members of the RTKIII family.15 The profile of 11 is summarized in Table 2. Our lead compound 11 had excellent selectivity over cKIT and cFMS (>100×), was clean on all tested subtypes of cytochrome p450 (>10 μM), and was potent in the cellular assay in the presence of 50% human plasma (25 nM). Subsequently, 11 was progressed to rat PK, which revealed that compound 11 suffered from poor oral exposure (16% F, dosed as a 20% Solutol aqueous solution, see Table 3).
Table 2. In Vitro Profile of Compounds 11 and 27–29a.
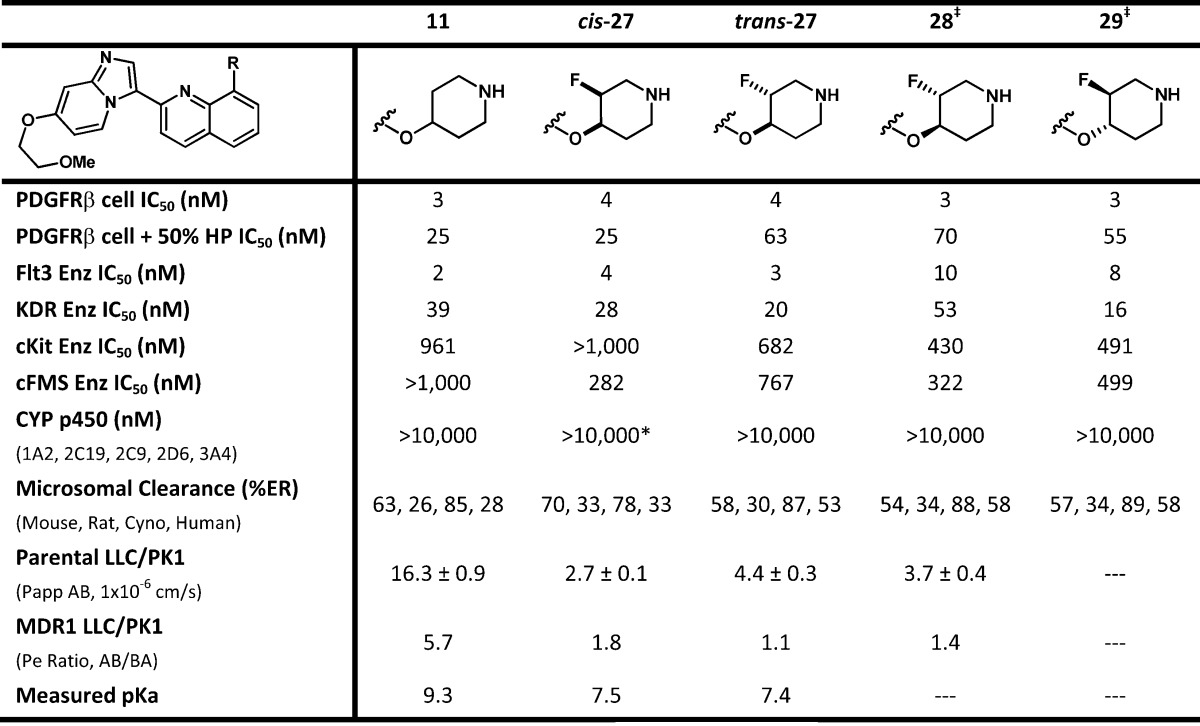
Key: *, compound cis-27 had an IC50 of 5880 nM on CYP 2D6; ‡, the absolute stereochemistry of 28 and 29 are arbitrarily assigned.
Table 3. Male Rat PK Profile of Compounds 11, 27, and 28.
| male rat PK | 11 | cis-27 | trans-27 | 28 | |
|---|---|---|---|---|---|
| 1 mg/kg IV | CL (mL/min/kg) | 33 | 28 | 24 | 20 |
| ER | 48 | 41 | 34 | 29 | |
| 10 mg/kg PO | AUC (μg)(hr)/mL | 0.80 | 0.73 | 2.1 | 2.3 |
| Cmax (μg/mL) | 0.07 | 0.07 | 0.12 | 0.16 | |
| % F | 16 | 13 | 30 | 28 |
These data, in combination with moderate IV clearance and excellent solubility (>1000 μg/mL) suggested that the poor oral exposure was not solely due to in vivo instability. Compound 11 was analyzed in a MDR1 cell line assay, and although attenuated apical recoveries were noted (50% recovery), it displayed high passive permeability along with efflux transport by P-glycoprotein (Pgp) (Pe ratio = 5.7).
The measured pKa of 11 was found to be 9.3, which facilitated good solubility but potentially played a role in Pgp efflux. By attenuating the pKa of the piperdine amine, we hoped to positively influence passive permeability19−22 and reduce Pgp mediated efflux.23−25 Attempts had been made earlier to reduce the pKa of 11 by employing a morpholine headgroup (20), installing an ether or hydroxyl on the ring (17, 18, and 22), moving the amine closer to the ether linkage (15 and 26) or installing an ester adjacent to the amine (23 and 24). Unfortunately, these attempts either decreased PDGFRβ potency or led to high predicted microsomal clearance.
Our focus then turned to the utility of fluorine. It is well precedented that introduction of a highly polarized and strong C–F bond (105 kcal/mol) can reduce the basicity of proximal amines.26,27 In addition, installation of fluorine not only creates a strong C–F bond but also leads to increases in the strength of adjacent C–O and C–C bonds.28,29 Therefore, the addition of fluorine on the piperidine ring had the potential to not only attenuate the amine basicity but also enhance metabolic stability.
Accordingly, we installed a single fluorine atom on the piperidine ring (Table 2), arriving at two racemic but diastereomerically pure compounds, cis-27 and trans-27. For cyclic alkyl amines, the decrease of pKa from an axial fluorine substituent can be considerably smaller than that seen for an equatorial fluorine.23−25 Therefore, the relative orientation of the fluorine substituent can have an effect on the pKa of the piperidine amine. On the basis of our experimental measurement, we found that the fluorine atom had attenuated the pKa nearly two units, from 9.3 for compound 11 to 7.4 for trans-27. Although we had anticipated a difference in the pKa of cis-27 and trans-27, they were essentially the same.
We anticipated that the observed decrease in pKa of the piperidine amine would be sufficient to increase the passive permeability of 11 while decreasing Pgp mediated efflux and lead to improved bioavailability.19−25 During our in vitro profiling of these compounds, we were surprised to find that installation of a fluorine atom on the piperidine ring had caused a reduction in the apparent permeability of both cis- and trans-27 when compared to compound 11 (albeit with attenuated apical recoveries of 30% for 27). This result is in contrast to our previous experience, as well as literature precedents that suggest a more neutral species (i.e., decreasing the pKa of a basic amine) will improve membrane permeability.19−25 Although the apparent permeability of cis-27 and trans-27 had been reduced, Pgp mediated efflux was significantly decreased, as seen in the MDR1 cell line assay, when compared to 11.
Both cis- and trans-27 were then progressed into rat pharmacokinetic studies. The results demonstrate that the installation of fluorine clearly reduced the IV clearance, from 33 (11) to 24 mL/min/kg (trans-27). Despite the improvement in IV clearance, cis-27 was unexpectedly indistinguishable from 11 in rat PK (Table 3). Conversely, trans-27 had significantly better in vivo exposure (2–3× improved AUC, Cmax, and % F) than cis-27 and 11. We were gratified to find that the installation of a single fluorine atom had not only aided in maintaining the desired potency and selectivity but provided a significant improvement in oral bioavailability.
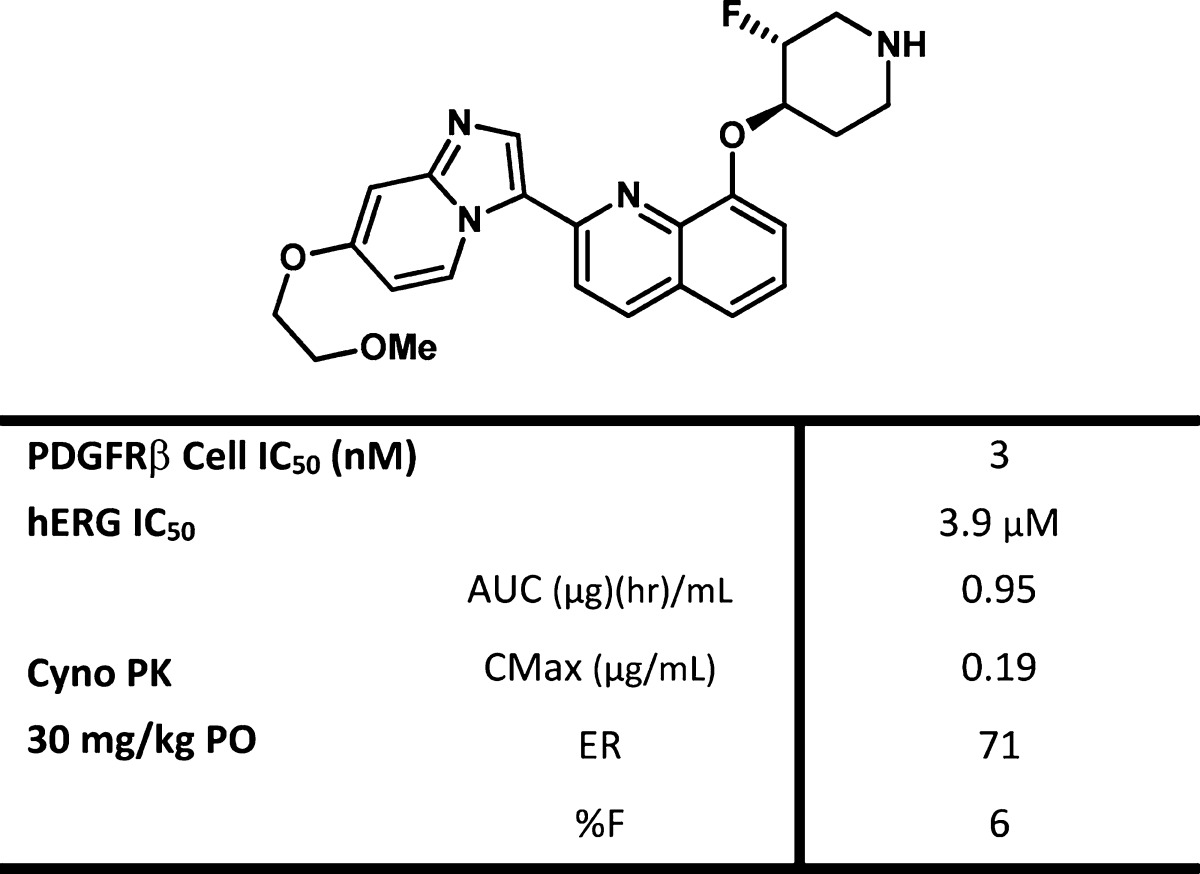
With the identification of trans-27, we separated and profiled the enantiomers 28 and 29, to determine if they would differentiate (the absolute stereochemistry shown for 28 and 29 are arbitrarily assigned). These two compounds were nearly identical in all assays with the exception of the KDR enzyme assay, in which 28 was 53 nM on KDR and 17× selective for PDGFRβ, versus the less selective 29, which was 16 nM on KDR and only 5× selective for PDGFRβ.15 When fluoro piperdine 28 was evaluated in vitro, it was found to maintain the desirable properties of 11 while improving upon 11 by lowering Pgp efflux. Oral exposure of 28 in rat was improved when compared to 11 with a concomitant decrease in IV clearance (see Table 3).
Compound 28 was also evaluated in a pharmacokinetic–pharmacodynamic (PKPD) assay, which we employed as a surrogate for the efficacy of our PDGFR inhibitors (Figure 3). The PKPD experiment was performed in female nude mice bearing subcutaneous C6 tumor xenografts. pPDGFR levels were determined and the values normalized to TotalERK by Western Blot. Compound 28 was evaluated at three different time points and at three doses. Following oral administration of 28 as a single dose, a PD response was observed in a dose-dependent manner. The effect of 28 was maximized at the 6 h time point with pPDGFR levels returning to those of the control after 12 h. On the basis of this data, the EC50 of 28 was determined to be a total drug concentration of 65 ng/mL.
Figure 3.

PKPD study of compound 28 as a single oral dose. Multiple time points and doses were evaluated. The ratio of pPDGFR/tERK was measured and compared to control.
Compound 28 was determined to be an excellent compound for the investigation of PDGFRβ inhibition in rodents and was well tolerated on chronic PO dosing in rats at doses of up to 50 mg/kg BID for two weeks. However, it suffered from high clearance in cynomolgus monkeys (see Table 4), which was predicted by the in vitro microsome stability assay of 28 (Table 2). The resulting lack of exposure in monkeys prevented tolerability assessment in second species and prohibited additional studies. Within this series we have established robust IV/IV correlation in both rat and monkeys. As these analogues consistently display high first pass clearance in second species, complicating preclinical toxicology assessment, we continued our medicinal chemistry efforts toward the development of a compound with improved oral exposure and decreased clearance in second species. These efforts will be addressed in a subsequent manuscript.
Table 4. hERG and Cyno PK Profile of 28.
Acknowledgments
We would like to thank Philip Anderson and Ben Colson for obtaining HRMS and pKa data, respectively.
Glossary
ABBREVIATIONS
- AUC
total area under the plasma drug concentration–time curve
- DBA(dba)
dibenzylideneacetone
- DCE
1,1-dichloroethane
- ER
extraction ratio
- HMDS
1,1,1,3,3,3-hexamethyldisilazane
- MDR1
multidrug resistance gene
- PDGFR
platlet-derived growth factor receptor
- Pgp
P-glycoprotein
- PK
pharmacokinetic
- pKa
logarithmic acid dissociation constant
- PKPD
pharmacokinetic–pharmacodynamic
- SAR
structure–activity relationship
- TFA
trifluoroacetic acid
- X-Phos
2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
Supporting Information Available
Protocol of the PDGFR cellular assay and pharmacokinetic/pharmacodynamic assay. Experimental procedures for the synthesis and chacterization of 3–11, 27, and 28. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Lewis N. L. The platelet-derived growth factor receptor as a therapeutic target. Curr. Oncol. Rep. 2007, 9, 89–95. [DOI] [PubMed] [Google Scholar]
- Board R.; Jayson G. C. Platelet-derived growth factor receptor (PDGFR): A target for anticancer therapeutics. Drug Resist. Updates 2005, 8, 75–83. [DOI] [PubMed] [Google Scholar]
- Wong N. A. C. S.; Mangwana S. KIT and PDGFRα mutational analyses of mixed cell-type gastrointestinal stromal tumors. Histopathology 2007, 51, 758–762. [DOI] [PubMed] [Google Scholar]
- Joyce J. A. Therapeutic targeting of the tumor microenvironment. Cancer Cell 2005, 7, 513–520. [DOI] [PubMed] [Google Scholar]
- Huang J.; Soffer S. Z.; Kim E. S.; McCrudden K. W.; Huang J.; New T.; Manley C. A.; Middlesworth W.; O’Toole K.; Yamashiro D. J.; Kandel J. Vascular remodeling marks tumors that recur during chronic suppressionof angiogenesis. J. Mol. Cancer Res. 2004, 2, 36–42. [PubMed] [Google Scholar]
- Bonner J. C.; Lindroos P. M.; Rice A. B.; Moomaw C. R.; Morgan D. L. Induction of PDGF receptor-α in rat myofibroblasts during pulmonary fibrogenesis in vivo. Am. J. Physiol.: Lung Cell. Mol. Physiol. 1998, 274, L72–L80. [DOI] [PubMed] [Google Scholar]
- Matsuno K.; Nakajima T.; Ichimura M.; Giese N. A.; Yu J.-C.; Lokker N. A.; Ushiki J.; Ide S.; Oda S.; Nomoto Y. Potent and selective inhibitors of PDGFR receptor phosphorylation. 2. Synthesis, structure activity relationship, improvement of aqueous solubility, and biological effects of 4-[4-(N-substituted(thio)carbamoyl)-1-piperazinyl]-6,7-dimethoxyquinazoline derivatives. J. Med. Chem. 2002, 45, 4513–4523. [DOI] [PubMed] [Google Scholar]
- Roberts W. G.; Whalen P. M.; Soderstrom E.; Moraski G.; Lyssikatos J. P.; Wang H.-F.; Cooper B.; Baker D. A.; Savage D.; Dalvie D.; Atherton J. A.; Ralston S.; Szewc R.; Kath J. C .; Lin J.; Soderstrom C.; Tkalcevic G.; Cohen B. D.; Pollack V.; Barth W.; Hungerford W.; Ung E. Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451. Cancer Res. 2005, 65, 957–966. [PubMed] [Google Scholar]
- For a more recent review see:Dai Y. Platelet-derived growth factor receptor kinase inhibitors: a review of the recent patent literature. Expert Opin. Ther. Patents 2010, 20, 885–907. [DOI] [PubMed] [Google Scholar]
- Robinson D. R.; Wu Y.-M.; Lin S.-F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–557. [DOI] [PubMed] [Google Scholar]
- Ullrich A.; Schlessinger J. Signal transduction by receptors with tyrosine activity. Cell 1990, 61, 203–212. [DOI] [PubMed] [Google Scholar]
- Kaipainen A.; Korhonen J.; Pajusola K.; Aprelikova O.; Persico M. G.; Terman B. I.; Alitalo K. The related FLT4, FLT1, and KDR receptor tyrosine kinases show distinct expression patterns in human endothelial cells. J. Exp. Med. 1993, 178, 2077–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Please see the Supporting Information to view key protein sequence variations that are within the active site.
- Allen S.; Greschuk J. M.; Kallan N. C.; Marmsater F. P.; Munson M. C.; Rizzi J. P.; Robinson J. E.; Schlachter S. P.; Topalov G. T.; Zhao Q.. Imidazo[1,2-A] Pyridine Compounds as Receptor Tyrosine Kinase Inhibitors. International Patent Application WO 2008/124323 A1, 2008.
- We experienced difficulties obtaining a reproducible PDGFR enzyme assay and chose to use a more robust cellular assay for the screening of our compounds.
- Huang X.; Buchwald S. L. New ammonia equivalents for the Pd-catalyzed amination of aryl halides. Org. Lett. 2001, 3, 3417–3419. [DOI] [PubMed] [Google Scholar]
- Touré B. B.; Lane B. S.; Sames D. Catalytic C–H arylation of SEM-protected azoles with palladium complexes of NHCs and phosphines. Org. Lett. 2006, 8, 1979–1982. [DOI] [PubMed] [Google Scholar]
- Koubachi J.; El Kazzouli S.; Berteina-Raboin S.; Mouaddib A.; Guillaumet G. Regioselective palladium-catalyzed arylation and heteroarylation of imidazo[1,2-a]pyridines. Synlett 2006, 19, 3237–3242. [Google Scholar]
- Faller B.; Urban L.; Mannhold R.. Hit and Lead Profiling: Identification and Optimization of Drug-Like Molecules; Wiley-VCH: Berlin, Germany, 2009. [Google Scholar]
- Kerns E. H.; Di L.. Drug-Like Properties: Concepts, Structure Design and Methods: from ADME to Toxicity Optimization; Academic Press: New York, 2008. [Google Scholar]
- Gleeson M. P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [DOI] [PubMed] [Google Scholar]
- Thomas V. H.; Bhattachar S.; Hitchingham L.; Zocharski P.; Naath M.; Surendran N.; Stoner C. L.; El-Kattan A. The road map to oral bioavailability: an industrial perspective. Expert Opin. Drug Metab. Toxicol. 2006, 2, 591–608. [DOI] [PubMed] [Google Scholar]
- Lerchner A.; Machauer R.; Betschart C.; Veenstra S.; Rueeger H.; McCarthy C.; Tintelnot-Blomley M.; Jaton A.-L.; Rabe S.; Desrayaud S.; Enz A.; Staufenbiel M.; Paganetti P.; Rondeau J.-M.; Neumann U. Macrocyclic BACE-1 inhibitors acutely reduce Aβ in brain after po application. Bioorg. Med. Chem. Lett. 2010, 20, 603–607. [DOI] [PubMed] [Google Scholar]
- Su D.-S.; Lim J. L.; Tinney E.; Wan B.-L.; Murphy K. L.; Reiss D. R.; Harrell C. M.; O’Malley S. S.; Ransom R. W.; Chang R. S. L.; Pettibone D. J.; Yu J.; Tang C.; Prueksaritanont T.; Freidinger R. M.; Bock M. G.; Anthony N. J. 2-Aminobenzophenones as a novel class of bradykinin B1 receptor antagonists. J. Med. Chem. 2008, 51, 3946–3952. [DOI] [PubMed] [Google Scholar]
- Cox C. D.; Breslin M. J.; Whitman D. B.; Coleman P. J.; Garbaccio R. M.; Fraley M. E.; Zrada M. M.; Buser C. A.; Walsh E. S.; Hamilton K.; Lobell R. B.; Tao W.; Abrams M. T.; South V. J.; Huber H. E.; Kohl N. E.; Hartman G. D. Kinesin spindle protein (KSP) inhibitors. Part V: Discovery of 2-propylamino-2,4-diaryl-2,5-dihydropyrroles as potent, water-soluable KSP inhibitors, and modulation of their basicity by b-fluorination to overcome cellular efflux by P-glycoprotein. Bioorg. Med. Chem. Lett. 2007, 17, 2697–2702. [DOI] [PubMed] [Google Scholar]
- Morgenthaler M.; Schweizer E.; Hoffmann-Roder A.; Benini F.; Martin R. E.; Jaeschke G.; Wagner B.; Fischer H.; Bendels S.; Zimmerli D.; Schneider J.; Diederich F.; Kansy M.; Muller K. Predicting and tuning physicochemical properties in lead optimization: Amine basicities. ChemMedChem 2007, 2, 1100–1115. [DOI] [PubMed] [Google Scholar]
- van Niel M. B.; Collins I.; Beer M. S.; Broughton H. B.; Cheng S. K. F.; Goodacre S. C.; Heald A.; Locker K. L.; MacLeod A. M.; Morrison D.; Moyes C. R.; O’Connor D.; Pike A.; Rowley M.; Russel M. G. N.; Sohal B.; Stanton J. A.; Thomas S.; Verrier H.; Watt A. P.; Castro J. L. Fluorination of 3-(3-(piperdin-1-yl)propyl)indoles and 3-(3-(piperazin-1-yl)propyl)indoles gives selective human 5-HT1D receptor ligands with improved pharmacokinetic profiles. J. Med. Chem. 1999, 42, 2087–2104. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.