

Abstract

The recently described synthetic GPR17 agonist 2-carboxy-4,6-dichloro-1H-indole-3-propionic acid (1) was prepared in tritium-labeled form by catalytic hydrogenation of the corresponding propenoic acid derivative 8 with tritium gas. The radioligand [3H]PSB-12150 (9) was obtained with a specific activity of 17 Ci/mmol (629 GBq/mmol). It showed specific and saturable binding to a single binding site in membrane preparations from Chinese hamster ovary cells recombinantly expressing the human GPR17. A competition assay procedure was established, which allows the determination of ligand binding affinities.
Keywords: MDL29,951; montelukast; multiple sclerosis; nucleotide; orphan GPCR; pranlukast; radioligand
The G protein-coupled receptor GPR17 is an orphan, rhodopsin-like receptor, which is phylogenetically related to nucleotide P2Y and cysteinyl-leukotriene receptors (Cys-LTR).1 It is predominantly expressed in the central nervous system (CNS), particularly in differentiating oligodendrocyte precursor cells.2−4 GPR17 was identified as a key player in the modulation of CNS myelination and has recently been shown to negatively regulate the oligodendrocyte differentiation process.2,5 Hence, inhibition of GPR17 emerges as a promising therapeutic approach for the treatment of demyelinating diseases such as multiple sclerosis (MS).
GPR17 has been reported to be activated by nucleotides (UDP) and nucleotide-sugars (UDP-glucose and UDP-galactose) as well as cysteinyl-leukotrienes (CysLTC4 and CysLTD4).6−10 Benned-Jensen and Rosenkilde confirmed an activation by UDP and UDP-glucose but failed to show GPR17 activation by cysteinyl-leukotrienes,11 whereas Maekawa et al.12 and Qi et al.13 found that neither nucleotides nor cysteinyl-leukotrienes were able to activate GPR17. Instead, GPR17 was identified as a negative regulator of the Cys-LT1R suppressing CysLT1R-mediated function at the cell membrane.12
Because of these contradictory results, the activation of GPR17 by nucleotides and cysteinyl-leukotrienes is still a subject of intense discussion and controversy.
Recently Hennen et al.5 identified 2-carboxy-4,6-dichloro-1H-indole-3-propionic acid (1, MDL29,951) and 3-[(E)-3-(anilino)-3-oxoprop-1-enyl]-4,6-dichloro-1H-indole-2-carboxylic acid (GV150526A, gavestinel, 2) as synthetic, small molecule GPR17 agonists and characterized them in different functional assays including Gq-mediated intracellular calcium mobilization, Gi-coupled inhibition of adenylate cyclase (AC), Gs-coupled stimulation of AC, and dynamic mass redistribution (Figure 1). Activation of GPR17 by 1 was demonstrated in multiple cell types including recombinant cells as well as primary rat oligodendrocytes. Compound 1 was found to selectively activate GPR17, but not P2Y or Cys-LT receptors. Very recently, several analogues of 1 were synthesized, and the first structure–activity relationships of these new GPR17 agonists have been described.14 Both indole derivatives, 1 and 2, had originally been developed as NMDA receptor antagonists.15,16
Figure 1.

Structure of synthetic GPR17 agonists.
Abbracchio and co-workers reported that the Cys-LTR antagonists pranlukast and montelukast, and the metabolically stable ATP analogue ATPγS acted as GPR17 antagonists.1,7 Recently, pranlukast was confirmed to inhibit GPR17 activation induced by the newly discovered synthetic agonist 1, while montelukast was found to be inactive.5
So far, no radioligand is available to determine GPR17 ligand binding affinities. Especially in view of the controversies in the field regarding GPR17 agonists and antagonists, such a tool would be highly useful to directly study receptor–ligand interaction. Thus, we decided to prepare the potent and selective GPR17 agonist 1 in tritium-labeled form.
As a suitable precursor we chose the corresponding unsaturated analogue 8, in which the propionic acid was formally replaced by a propenoic acid residue. The double bond in the side chain was expected to be selectively hydrogenated under mild conditions in the presence of chlorine substituents on the aromatic ring and without hydrogenation of the aromatic double bonds.
The radioligand 9 was prepared as depicted in Scheme 1. 3,5-Dichloroaniline (3) was converted to ethyl-4,6-dichloro-1H-indole-2-carboxylate (5) in a two step synthetic procedure by Japp-Klingemann condensation followed by Fischer indole (aza-Cope) ring closure reaction. Vilsmeier–Haack formylation of 5 according to Baron et al.17 afforded aldehyde 6. Compound 6 was subsequently reacted with ethyl (triphenylphosphoranylidene)acetate in a Horner–Wadsworth–Emmons reaction yielding alkene 7.18 Subsequent ester hydrolysis afforded the desired precursor molecule 3-(2-carboxyvinyl)-4,6-dichloro-1H-indole-2-carboxylic acid (8, for experimental details and NMR spectra see Supporting Information). Radiolabeling of 8 was accomplished by catalytic hydrogenation with tritium gas (custom-labeling performed by Quotient Bioscience, U.K.). The reaction was performed in tetrahydrofuran (THF) catalyzed by palladium on activated charcoal. The radioactively labeled product 9 was obtained in a specific activity of 17 Ci/mmol (629 GBq/mmol) and with a radiochemical purity of 99.4% (Supporting Information Figure S2). The moderate specific activity may be explained by possible enolization of the enone structure.
Scheme 1. Preparation of the Radioligand 9.

Reagents and conditions: (a) HCl, NaNO2, KOH, ethyl 2-methylacetoacetate, EtOH, 0 °C, 20 min, then 40 °C, 15 min; (b) polyphosphoric acid, toluene, 45 °C, 20 min; (c) DMF, POCl3, CH2Cl2, reflux, 2.5 h; (d) argon, ethyl (triphenylphosphoranylidene)acetate, toluene, reflux, overnight; (e) THF/H2O (1:1), LiOH·H2O, rt, overnight; (f) THF, [3H]H2, Pd/C.
For radioligand binding studies, membrane preparations of a Chinese hamster ovary (CHO) cell line stably transfected with the human GPR17 (CHO-hGPR17) by an inducible Flp-In system (Flp-In T-Rex) to obtain high expression levels were used.5,19 As an additional cell line 1321N1 astrocytoma cells stably transfected with the human GPR17 by means of a retroviral vector (pLXSN) were available.5 Quantitative real-time PCR analyses revealed an approximately 20-fold higher mRNA expression for GPR17 in case of the inducible (doxycycline 1 μg/mL) Flp-In T-Rex CHO cell line stably expressing the human GPR17 (CHO-hGPR17) than for the recombinant 1321N1 astrocytoma cell line (Supporting Information Figure S3).
Specific [3H]PSB-12150 binding was observed to the recombinant cells, while the radioligand showed only negligible binding to nontransfected CHO cells under the same conditions (Figure 2). Initially, the effects of the buffer composition on specific [3H]PSB-12150 binding were investigated. The various commonly used buffers, such as phosphate-buffered saline (PBS), (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and tris(hydroxylmethyl)aminomethane (TRIS) gave similar results. The addition of 100 mM NaCl led to a decrease in specific binding, whereas in the presence of 10 mM MgCl2, specific [3H]PSB-12150 binding was significantly increased. It is well-known that sodium ions can act as negative allosteric modulators of many GPCRs, and its binding site has recently been elucidated in adenosine receptors.20 In contrast, Mg2+ ions may enhance the formation of a high-affinity agonist–receptor–G protein complex acting as a positive allosteric modulator, and the same effect has recently been observed with an agonist radioligand at GPR35.21 Thus, 50 mM TRIS buffer, pH 7.4, containing 10 mM MgCl2 was found to be a suitable buffer for performing radioligand binding studies with [3H]PSB-12150, and it was therefore used in all further experiments.
Figure 2.
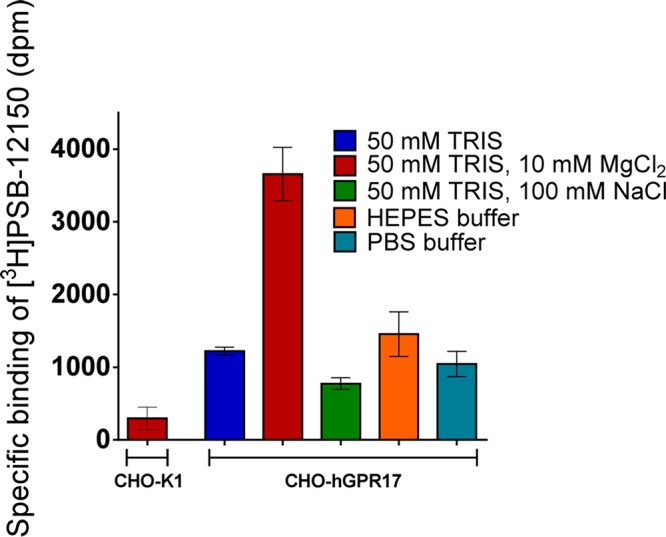
Specific binding of [3H]PSB-12150 to CHO-K1 and CHO-hGPR17 membrane preparations and effects of different buffers and ions on specific binding. The experiments were performed at 25 °C (60 min) using 25 nM [3H]PSB-12150 and 30 μg of the respective membrane preparation.
As a next step, kinetic studies with CHO-hGPR17 cell membranes were performed using 25 nM [3H]PSB-12150 in a total volume of 1 mL of 50 mM TRIS buffer, pH 7.4, supplemented with 10 mM MgCl2 in the presence of 30 μg protein. Both association and dissociation appeared monophasic (Figure 3). The radioligand showed fast association (determined at 25 °C) and equilibrium was reached after less than 60 min and remained stable for at least 4 h. The binding was rapidly reversed after the addition of 100 μM of unlabeled 1 (Figure 3B). As shown in Figure 3A, t1/2 was 1.8 min, indicating that an incubation time of 60 min is sufficient to ensure equilibrium binding of [3H]PSB-12150.
Figure 3.

Kinetics of [3H]PSB-12150 binding (25 nM) to membranes of CHO cells recombinantly expressing human GPR17 receptors at 25 °C with 30 μg protein/vial in 50 mM TRIS buffer, pH 7.4, 10 mM MgCl2: (A) association curve; (B) dissociation curve; dissociation was achieved by the addition of 100 μM compound 1 after 1 h of preincubation. Data points represent means ± SEM of at least two independent experiments, each performed in duplicates.
As depicted in Figure 4 saturation experiments in membranes of CHO cells with high expression of GPR17 using 13 different radioligand concentrations ranging from 12.5 to 10 000 nM revealed a KD value of 1256 nM and an apparent Bmax value of 56.6 pmol/mg protein. Despite the relatively high KD value it was possible to perform highly reproducible binding assays with [3H]PSB-12150 provided that ice-cold wash buffer and a cell line with a high GPR17 expression level were used. As stated by Hulme et al. the use of ice-cold wash buffer can extend the usable range of radioligands by up to 100-fold since the Koff is often greatly reduced at low temperature.22 Filtration through glass fiber GF/B filters (1 μm pores) was found to be suitable for the utilized membrane preparation as previously shown for similar preparations containing other GPCRs.21,23−26 The washing procedure of the filters was carefully checked, and 2 to 3 times washing was found to provide consistent results with relatively high specific binding, whereas additional washing steps caused dissociation of the radioligand (see Supporting Information Figure S4).
Figure 4.

(A) Saturation curve for specific [3H]PSB-12150 binding to membranes of CHO cells recombinantly expressing the human GPR17. Saturation experiments were performed at 25 °C, incubation for 60 min, with 50 μg of protein per vial in 50 mM TRIS buffer, pH 7.4, containing 10 mM MgCl2. Data points represent means ± SEM of two independent experiments, each performed in duplicates. The following binding parameters were calculated: KD = 1256 ± 138 nM and Bmax = 56.6 ± 5.8 pmol/mg protein. (B) Scatchard plot of the same data.
Specific binding was found to be protein-dependent: it increased with increasing amounts of protein (see Supporting Information Figure S5). In our recombinant 1321N1 astrocytoma cell line, which expresses a moderate level of human GPR17, we could only detect a much lower degree of specific binding than in CHO-hGPR17 cells, as expected (see Supporting Information Figure S5). Thus, that cell line is not as well suitable for radioligand binding assays with [3H]PSB-12150.
A radioligand concentration of 25 nM [3H]PSB-12150 was found to be suitable for performing competition binding assays. The nonspecific binding at that concentration amounted to 36% of total binding and was thus in a tolerable range. Homologous competition experiments with the unlabeled ligand 1 were performed using either a high concentration of 1 (100 μM) or a high concentration of the antagonist pranlukast (100 μM) to determine nonspecific binding. Both compounds gave the same percentage of nonspecific binding. Moreover, homologous competition assays for compound 1 resulted in nearly identical Ki values of 2.32 μM (using 1 for the determination of nonspecific binding, Figure 5A) and 1.21 μM (using pranlukast for nonspecific binding, Figure 5B), which are in good agreement with the KD value determined for the radioligand.
Figure 5.
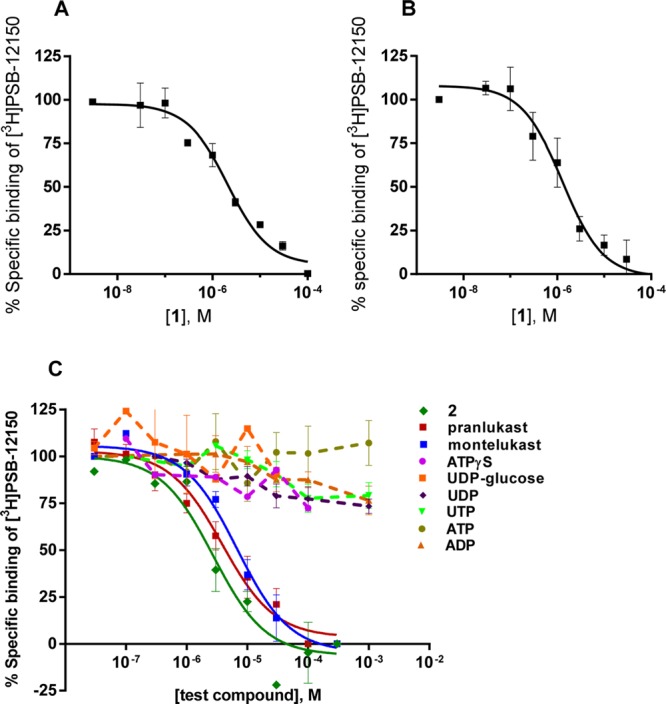
Competition experiments at membrane preparations from recombinant CHO cells expressing human GPR17. The experiments were performed at 25 °C (60 min) using 25 nM [3H]PSB-12150 and 50 μg protein/vial in 50 mM TRIS buffer, pH 7.4, 10 mM MgCl2. (A) Homologous competition experiment. Compound 1 was used for the determination of nonspecific binding. A Ki value of 2.32 ± 0.32 μM was determined for compound 1. (B) Homologous competition experiment. Pranlukast was used for the determination of nonspecific binding. A Ki value of 1.21 ± 0.33 μM was determined for compound 1. (C) Heterologous competition experiments. Ki values were calculated as follows: 1.63 ± 0.85 μM for compound 2, 4.06 ± 0.82 μM for pranlukast, and 6.54 ± 0.78 μM for montelukast, respectively. Data points represent means ± SEM from 2 to 4 independent experiments, each performed in duplicates.
As a next step, several ligands, putative agonists, and antagonists were investigated in heterologous competition experiments. None of the tested nucleotides (ATP, ADP, UTP, and UDP) showed any inhibition or modulation of [3H]PSB-12150 binding at concentrations up to 1 mM. Likewise, the nucleotide-sugar UDP-glucose did not displace [3H]PSB-12150 from its binding site (Figure 5 and Supporting Information Figure S6). However, as expected, the nucleotide GTP inhibited [3H]PSB-12150 binding at a concentration of 100 μM almost completely (Supporting Information Figure S5); this can be explained by its binding to the G protein, which results in its uncoupling from the receptor resulting in a negative allosteric modulation of agonist binding.27,28 Cysteinyl-leukotriene C4 did not show any significant effect on [3H]PSB-12150 binding at concentrations up to 1 μM (Supporting Information Figure S6). In contrast, indole derivative 2 (Figure 1) showed a concentration-dependent inhibition of [3H]PSB-12150 with a Ki value of 1.63 μM. Thus, it displayed a similarly high affinity to GPR17 as 1. In a previous study, 2 had shown lower potency than 1 in dynamic mass redistribution assays. The discrepancy in binding and functional results might indicate that 2 is a partial GPR17 agonist.
Furthermore, putative antagonists were investigated. The Cys-LTR antagonists pranlukast and montelukast were both able to displace the radioligand, and similar affinity was determined for both compounds: a Ki value of 4.06 μM for pranlukast and of 6.54 μM for montelukast. While pranlukast had previously been found to block GPR17 in a functional assay, reports on montelukast were contradictory.1,5 One likely explanation would be that promiscuous compounds may show additional effects in functional assays due to interaction with natively expressed receptors and thereby offset the GPR17-mediated effect. These results show that radioligand binding studies are crucial for studying receptor–ligand interaction of (potentially) nonselective compounds. Unbiased ligand binding data are particularly valuable in drug development for structure–activity relationship analysis and for optimization of lead structures.
In the newly developed radioligand binding studies, we could confirm that the Cys-LTR antagonists bind to GPR17, although with only moderate affinities. However, we did not observe any binding affinity or modulation of radioligand binding by the nucleotides UDP and UDP-glucose and of the cysteinylleukotriene C4, which were previously postulated to be GPR17 agonists. Our results suggest that these nucleotides might not be the endogenous agonists of GPR17. We cannot exclude, however, that uracil nucleotides and nucleotide-sugars bind to a different binding site than our radioligand [3H]PSB-12150. On the basis of molecular modeling studies applying docking and molecular dynamics simulations (MD), it had been speculated that two different binding sites are present on GPR17.6 However, at least some modulation might be expected if two different agonists would simultaneously bind to the same receptor. The current data are therefore not supporting an interaction of nucleotides and nucleotide-sugars with GPR17.
In conclusion, we developed the first radioligand, [3H]PSB-12150, for GPR17 and characterized its binding properties. In addition, we investigated the binding affinities of previously reported agonists and antagonists. Since GPR17 has been postulated to be a promising target for the therapy of demyelinating diseases such as multiple sclerosis, brain and spinal cord injury, and neurodegenerative diseases, GPR17 ligands, in particular antagonists, represent potential new drug candidates. Even though [3H]PSB-12150 exhibits only moderate binding affinity for GPR17, a highly reproducible radioligand binding assay procedure was developed, which should be well suitable for compound screening. The new GPR17 radioligand represents a valuable tool for studying GPR17 ligands, and in particular for the discovery of novel, more potent GPR17 agonists and antagonists as well as their optimization.
Acknowledgments
The authors thank Ms. Inge Renner for expert technical assistance.
Supporting Information Available
Synthetic procedures, analytical data, assay procedures, RT-PCR, and additional radioligand binding data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ These authors (M.K. and K.R.) contributed equally to this work.
This study was funded by a grant from the German Federal Ministry for Education and Research (BMBF) within the BioPharma initiative “Neuroallianz”.
The authors declare no competing financial interest.
Supplementary Material
References
- Ciana P.; Fumagalli M.; Trincavelli M. L.; Verderio C.; Rosa P.; Lecca D.; Ferrario S.; Parravicini C.; Capra V.; Gelosa P.; Guerrini U.; Belcredito S.; Cimino M.; Sironi L.; Tremoli E.; Rovati G. E.; Martini C.; Abbracchio M P. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 2006, 25, 4615–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Wu H.; Wang S.; Koito H.; Li J.; Ye F.; Hoang J.; Escobar S. S.; Gow A.; Arnett H. A.; Trapp B. D.; Karandikar N. J.; Hsieh J.; Lu Q. R. The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat. Neurosci. 2009, 12, 1398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecca D.; Trincavelli M. L.; Gelosa P.; Sironi L.; Ciana P.; Fumagalli M.; Villa G.; Verderio C.; Grumelli C.; Guerrini U.; Tremoli E.; Rosa P.; Cuboni S.; Martini C.; Buffo A.; Cimino M.; Abbracchio M. P. The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PLoS One 2008, 3, e3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M.; Daniele S.; Lecca D.; Lee P. R.; Parravicini C.; Fields R. D.; Rosa P.; Antonucci F.; Verderio C.; Trincavelli M. L.; Bramanti P.; Martini C.; Abbracchio M. P. Phenotypic changes, signaling pathway, and functional correlates of GPR17-expressing neural precursor cells during oligodendrocyte differentiation. J. Biol. Chem. 2011, 286, 10593–10604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennen S.; Peters L.; Wang H.; Spinrath A.; Merten N.; Blättermann S.; Akkari R.; Schrage R.; Simon K.; Schröder R.; Schulz D.; Vermeiren C.; Zimmermann K.; Kehraus S.; Drewke C.; Pfeifer A.; König G. M.; Mohr K.; Gillard M.; Müller C. E.; Lu Q. R.; Gomeza J.; Kostenis E. Decoding signaling and function of the orphan G protein-coupled receptor GPR17 with a small molecule agonist. Sci. Signaling 2013, 6, ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parravicini C.; Abbracchio M. P.; Fantucci P.; Ranghino G. Forced unbinding of GPR17 ligands from wild type and R255I mutant receptor models through a computational approach. BMC Struct. Biol. 2010, 10, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese A. M.; Trincavelli M. L.; Lecca D.; Coppi E.; Fumagalli M.; Ferrario S.; Failli P.; Daniele S.; Martini C.; Pedata F.; Abbracchio M. P. Functional characterization of two isoforms of the P2Y-like receptor GPR17: [35S]GTPgammaS binding and electrophysiological studies in 1321N1 cells. Am. J. Physiol., Cell Physiol. 2009, 297, C1028–1040. [DOI] [PubMed] [Google Scholar]
- Buccioni M.; Marucci G.; Dal B. D.; Giacobbe D.; Lambertucci C.; Soverchia L.; Thomas A.; Volpini R.; Cristalli G. Innovative functional cAMP assay for studying G protein-coupled receptors: application to the pharmacological characterization of GPR17. Purinergic Signalling 2011, 7, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniele S.; Trincavelli M. L.; Gabelloni P.; Lecca D.; Rosa P.; Abbracchio M. P.; Martini C. Agonist-induced desensitization/resensitization of human G protein-coupled receptor 17: a functional cross-talk between purinergic and cysteinyl-leukotriene ligands. J. Pharmacol. Exp. Ther. 2011, 338, 559–567. [DOI] [PubMed] [Google Scholar]
- Coppi E.; Maraula G.; Fumagalli M.; Failli P.; Cellai L.; Bonfanti E.; Mazzoni L.; Coppini R.; Abbracchio M. P.; Pedata F.; Pugliese A. M. UDP-glucose enhances outward K(+) currents necessary for cell differentiation and stimulates cell migration by activating the GPR17 receptor in oligodendrocyte precursors. Glia 2013, 61, 1155–1171. [DOI] [PubMed] [Google Scholar]
- Benned-Jensen T.; Rosenkilde M. M. Distinct expression and ligand-binding profiles of two constitutively active GPR17 splice variants. Br. J. Pharmacol. 2010, 159, 1092–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa A.; Balestrieri B.; Austen K. F.; Kanaoka Y. GPR17 is a negative regulator of the cysteinyl leukotriene 1 receptor response to leukotriene D4. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 11685–11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi A.-D.; Harden T. K.; Nicholas R. A. Is GPR17 a P2Y/ leukotriene receptor? Examination of uracil nucleotides, nucleotide sugars, and cysteinyl-leukotrienes as agonists of GPR17. J. Pharmacol. Exp. Ther. 2013, 347, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baqi Y.; Alshaibani S.; Ritter K.; Abdelrahman A.; Spinrath A.; Kostenis E.; Müller C. E. Improved synthesis of 4-/6-substituted 2-carboxy-1H-indole-3-propionic acid derivatives and structure–activity relationships as GPR17 agonists. MedChemComm 2014, 5, 86–92. [Google Scholar]
- Di Fabio R.; Capelli A. M.; Conti N.; Cugola A.; Donati D.; Feriani A.; Gastaldi P.; Gaviraghi G.; Hewkin C. T.; Micheli F.; Missio A.; Mugnaini M.; Pecunioso A.; Quaglia A. M.; Ratti E.; Rossi L.; Tedesco G.; Trist D. G.; Reggiani A. Substituted indole-2-carboxylates as in vivo potent antagonists acting as the strychnine-insensitive glycine binding site. J. Med. Chem. 1997, 40, 841–50. [DOI] [PubMed] [Google Scholar]
- Salituro F. G.; Harrison B. L.; Baron B. M.; Nyce P. L.; Stewart K. T.; Kehne J. H.; White H. S.; McDonald I. A. 3-(2-Carboxyindol-3-yl)propionic acid-based antagonists of the N-methyl-d-aspartic acid receptor associated glycine binding site. J. Med. Chem. 1992, 35, 1791–1799. [DOI] [PubMed] [Google Scholar]
- Baron B. M.; Cregge R. J.; Farr R. A.; Friedrich D.; Gross R. S.; Harrison B. L.; Janowick D. A.; Matthews D.; McCloskey T. C.; Meikrantz S.; Nyce P. L.; Vaz R.; Metz W. A. CoMFA, synthesis, and pharmacological evaluation of (E)-3-(2-carboxy-2-arylvinyl)-4,6-dichloro-1H-indole-2-carboxylic acids: 3-[2-(3-aminophenyl)-2-carboxyvinyl)-4,6-dichloro-1H-indole-2-carboxylic acid, a potent selective glycine-site NMDA receptor antagonist. J. Med. Chem. 2005, 48, 995–1018. [DOI] [PubMed] [Google Scholar]
- Bigge C. F.; Graham J.; Po-Wai Y.. Derivatives of 2-carboxyindoles having pharmaceutical activity. Warner Lambert Company, Ann Arbor, Michigan, USA, US Patent 5,284,862, 1994.
- Ward R. J.; Alvarez-Curto E.; Milligan G. Using the Flp-In T-Rex system to regulate GPCR expression. Methods Mol. Biol. 2011, 746, 21–37. [DOI] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; IJzerman A. P.; Cherzov V.; Stevens R. C. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm D.; Funke M.; Meyer A.; Müller C E. 6-Bromo-8-(4-[3H]methoxy-benzamido)-4-oxo-4H-chromene-2-carboxylic acid ([3H]PSB-13253): a powerful tool for studying orphan G protein-coupled receptor GPR35. J. Med. Chem. 2013, 56, 7084–7099. [DOI] [PubMed] [Google Scholar]
- Hulme E. C.; Trevethick M. A. Ligand binding assays at equilibrium: validation and interpretation. Br. J. Pharmacol. 2010, 161, 1219–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrmann T.; Hinz S.; Bertarelli D. C.; Li W.; Florin N. C.; Scheiff A. B.; Müller C. E. 1-Alkyl-8-(piperazine-1-sulfonyl) phenylxanthines: development and characterization of adenosine A2B receptor antagonists and a new radioligand with subnanomolar affinity and subtype specificity. J. Med. Chem. 2009, 52, 3994–4006. [DOI] [PubMed] [Google Scholar]
- Bertarelli D. C.; Diekmann M.; Hayallah A. M.; Rüsing D.; Iqbal J.; Preiss B.; Verspohl E. J.; Müller C. E. Characterization of human and rodent native and recombinant adenosine A2B receptors by radioligand binding studies. Purinergic Signalling 2006, 2, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller C. E.; Maurinsh J.; Sauer R. Binding of [3H]MSX-2 (3-(3-hydroxypropyl)-7-methyl-8-(m-methoxystyryl)-1-propargylxanthine) to rat striatal membranes: a new, selective antagonist radioligand for A2A adenosine receptors. Eur. J. Pharm. Sci. 2000, 10, 259–265. [DOI] [PubMed] [Google Scholar]
- Schiedel A. C.; Meyer H.; Alsdorf B. B.; Gorzalka S.; Brüssel H.; Müller C. E. [3H]Adenine is a suitable radioligand for the labeling of G protein-coupled adenine receptors but shows high affinity to bacterial contaminations in buffer solutions. Purinergic Signalling 2007, 3, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A.; Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 2002, 54, 323–373. [DOI] [PubMed] [Google Scholar]
- Müller C. E.; Schiedel A. C.; Baqi Y. Allosteric modulators of rhodopsin-like G protein-coupled receptors: opportunities in drug development. Pharmacol. Ther. 2012, 135, 292–315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.