

Abstract
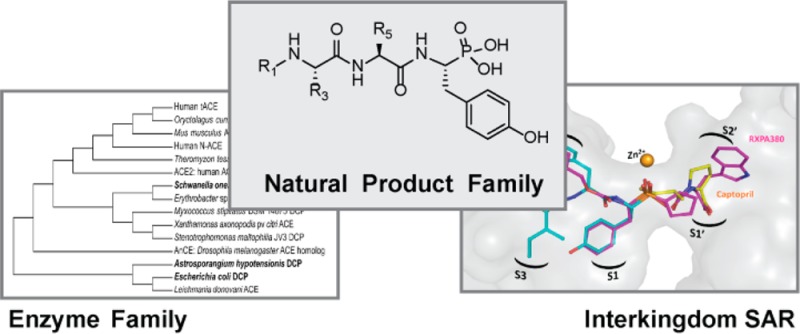
The K-26 family of bacterial secondary metabolites are N-modified tripeptides terminated by an unusual phosphonate analog of tyrosine. These natural products, produced via three different actinomycetales, are potent inhibitors of human angiotensin-I converting enzyme (ACE). Herein we investigate the interkingdom pharmacology of the K-26 family by synthesizing these metabolites and assessing their potency as inhibitors of both the N-terminal and C-terminal domains of human ACE. In most cases, selectivity for the C-terminal domain of ACE is displayed. Co-crystallization of K-26 in both domains of human ACE reveals the structural basis of the potent inhibition and has shown an unusual binding motif that may guide future design of domain-selective inhibitors. Finally, the activity of K-26 is assayed against a cohort of microbially produced ACE relatives. In contrast to the synthetic ACE inhibitor captopril, which demonstrates broad interkingdom inhibition of ACE-like enzymes, K-26 selectively targets the eukaryotic family.
Keywords: Angiotensin-I converting enzyme (ACE), K-26, bacterial dicarboxypeptidase, ACE inhibitor
A significant number of secondary metabolites produced by bacteria are potent and selective inhibitors of enzymes and receptor targets in organisms classified in other kingdoms.1 Whether these activities are the result of adaptive evolutionary pressures or coincidence is unknown, but increasing evidence is accumulating that microbial secondary metabolites play central roles in interkingdom chemical ecological relationships.2 The significance of this interkingdom pharmacology is also evident in the role of bacterial natural products in clinically approved therapies. A striking example of such interkingdom activity is the microbial natural products, K-26 (1) and related compounds (2–7), which are produced by actinomycetes isolated from soil-based ecosystems3−6 (Figure 1). These tripeptides contain a conserved terminal phosphonic acid analog of tyrosine, (R)-1-amino-2-(4-hydroxyphenyl)ethylphosphonic acid ((R)-AHEP, 8). This nonproteinogenic amino acid, unique to this family, is the zinc-binding pharmacophore that renders these compounds among the most potent reported natural product inhibitors of human angiotensin-I converting enzyme (ACE). ACE is a central player in mammalian physiology and a clinical target for the modulation of blood pressure via the renin angiotensin aldosterone system. The potency of the K-26 family against ACE, produced by a soil microbe for unknown purposes, raises questions pertaining to the structural origin of the potency and selectivity within the K-26 family and the chemical ecological roles of these compounds across kingdoms.
Figure 1.

K-26 analogs of interest in this study. K-26 (1), K-4 (2), 15-B-2 (3), SF2513 B (4), SF2513 C (5), and SF2513 A (6) have been isolated from actinomycete cultures and noted for their potent ACE inhibitory activity. SF2513 D (7) is a synthetic analog used as a replacement for SF2513A in this study. All analogs are characterized by a nonproteogenic phosphonate analog of tyrosine, 1-amino-2-(4-hydroxyphenyl)ethylphosphonic acid (AHEP) (8).
The importance of ACE inhibitors to human health has been emphasized by a World Health Organization World Health Statistics 2012 report. Hypertension is directly responsible for 12.8% of deaths worldwide and is a high-risk condition for stroke and coronary heart disease.7 ACE inhibitors were successfully developed based on a paradigm of rational drug design in the 1970s. However, current ACE-targeting therapies for hypertension are not without deleterious side effects such as cough and angioedema,8 which are likely a consequence of elevated levels of bradykinin and substance P,9,10 along with off-target effects of ACE inhibitors on tissue specific homologues of ACE and other carboxypeptidases distributed throughout mammalian cell biology. It was only after the discovery of the first generation ACE inhibitors that human somatic ACE (sACE) was shown to comprise two tandem homologous domains, the N- and the C- domain, each with an active site consisting of an HEXXH zinc binding sequence.11,12 These domains differ in pharmacological inhibitor preference and effectiveness at binding and cleaving biologically relevant substrates.13,14 Furthermore, a tissue-specific isoform, testicular ACE (tACE), has one active site and a single domain which, aside from a unique N-terminal leader sequence, is identical to the C-domain of sACE.15 Correspondingly, domain-selective inhibitors have potential as biochemical and physiological probes of ACE isoform function, and as improved therapies for hypertension.
Here we investigate the structural basis for K-26 inhibition of human ACE and the domain selectivity within the K-26 family of secondary metabolites. Chemical synthesis of five naturally produced structural variants of K-26 and analysis of their domain-selective inhibition properties facilitates the determination of the structure–activity relationships within the natural product family. The cocrystallization of the most potent member K-26, in both N- and C-domains, provides the first structural data of a natural product inhibitor in human ACE and reveals a new mode of inhibitor binding. To investigate interkingdom structure–activity relationships, we also explore the activity of K-26 and synthetic ACE inhibitors against a panel of bacterial zinc metalloenzyme analogs of human ACE. Structural departures from the K-26/hACE inhibitor complex, engendered either within the inhibitor or the enzyme, resulted in significantly decreased binding affinity, confirming previous studies.16 Understanding the biochemical and structural basis for K-26 inhibition provides an avenue for the future development of domain-selective inhibitors for improved ACE-targeting therapies and the groundwork for subsequent studies to elucidate the biological mechanism for the selectivity of K-26 for a mammalian enzyme.
Members of the K-26 family of natural products were originally discovered using bioassay guided fractionation of crude extracts via mammalian ACE inhibition. The tripeptide K-26 is produced by Astrosporangium hypotensionis (NRRL 2379),5,17 the SF2513 series of characterized analogs are produced by Streptosporangium nondiastiaticum SF2513,3 and K-4 and 15-B-2 are produced by Actinomadura spiculosospora,4,6 all of which are genera within the taxonomically diverse Streptosporangineae suborder of soil actinobacteria. With the exception of A. hypotensionis, the producing organisms are not publically available, and K-26 metabolites are generated at very low production levels. The full complement of the six known naturally occurring K-26 variants were synthesized, with the exception of the compound SF2513 A (6) (Supporting Information Scheme 1A), which was not readily accessible to us using an array of standard peptide coupling conditions (including HATU, PyBrop, EDC, and through generation of the acid chloride). Therefore, a close structural analog of SF2513 A (6) was generated lacking methylation on the penultimate amide nitrogen, and designated SF2513 D (7) (Supporting Information Scheme 1B). Both synthetic schemes yielded the desired K-26 analogs as mixtures of two major diastereomers, which were separated by preparative C18-HPLC. Comparison of 1H and 13C NMR acquired from synthetic (natural diastereomeric) natural products was consistent with extant literature spectral data, confirming the originally proposed structures and stereochemistries of these natural compounds.
Chemically synthesized K-26 family phosponotripeptides were initially assayed for mammalian ACE inhibition using the benchmark chromogenic substrate furylacryloyl-phenylalanyl-glycyl-glycine (FAPGG) and rabbit lung somatic ACE extract16,18 (Table 1). Consistent with previous data from K-26 SAR studies, diastereomers terminated by the naturally occurring (R)-AHEP amino acid were 10- to 400-fold more potent than those terminated by (S)-AHEP (Supporting Information Table 1). With the exception of SF2513 D (desmethyl SF2513 A), all synthesized natural products were potent nanomolar inhibitors of somatic rabbit lung ACE. SF2513 A was previously reported to be a nanomolar inhibitor of ACE, with the initial structure of SF2513 A being elucidated using 1H NMR, 13C NMR, and chemical degradation.3 With these results in mind, if the originally published structure of SF2513 A is correct, the N-methyl substituent must have an important role in promoting binding of SF2513 A in the active site of ACE. In general, N-acetylated family members were markedly more potent than their respective N-methylated congeners using the substrate FAPGG. Of the N-acetylated variants, K-26 was found to be the most potent inhibitor of rabbit lung somatic ACE, with an IC50 of 25 nM, followed by SF2513 C and then SF2513 B.
Table 1. ACE Inhibitory Activity of Synthesized K-26 Variants.
| IC50 data (nM) |
Ki data (nM) |
||||
|---|---|---|---|---|---|
| compd | sACEa | C-domb | N-domb | C-domb | N-domb |
| K-26 | 25d | 11d | 110e | 11f | 75d |
| K-4 | 370e | 590d | 220c | NTg | NT |
| 15-B-2 | 140e | 54e | 52c | NT | NT |
| SF2513 B | 100e | 12d | 76c | 21e | 33d |
| SF2513 C | 35e | 7e | 88e | 12f | 170d |
| SF2513 D | 5200d | NT | NT | NT | NT |
Rabbit lung sACE was tested for inhibition with the substrate FAPGG.
Z-FHL was used as a substrate with purified N- and C- domain ACE constructs. Standard deviations, reported in the Supporting Information on pp S89–S93, are as follows:
1–10%.
10–25%.
25–50%.
50–75% of the mean.
NT is not tested.
Each of the synthesized natural products that exhibited nanomolar somatic ACE inhibition were also tested for domain selectivity by measuring the IC50 of inhibition using the substrate Cbz-Phe-His-Leu-OH (Z-FHL)19−21 with purified N-domain and C-domain human ACE constructs. Analogs showing promising domain selectivity were reevaluated via measurement of the inhibition constant Ki. The N-methylated analogs, K-4 and 15-B-2, did not exhibit a large degree of domain-selectivity, and SF2513 B only showed a slight preference for the C-domain, whereas N-acetylated analogs K-26 and SF2513 C showed approximately 7- and 15-fold higher affinities for the C-domain, respectively.
To further understand the structural basis for inhibition of human ACE by the most potent member of this family of bacterial natural products, K-26 was separately cocrystallized in the N-domain and the C-domain. As shown in the resulting crystal structures, K-26 adopts a similar conformation in both domains (Figure 2A and B), conserving nearly identical potential hydrogen bond networks between the two domains (Supporting Information Table 2). K-26 has a unique binding motif, as it occupies the “non-prime” binding pockets of this mammalian ACE construct (Figure 3). The phosphonate coordinates the zinc, the side chain of the AHEP sits in the S1 binding pocket, the tyrosine side chain fills the S2 binding pocket, and the N-acetyl isoleucine occupies a S3 binding pocket. Other known inhibitors of the enzyme, including the clinically relevant captopril and lisinopril, and the synthetic phosphinic inhibitor RXPA380, fill the “prime” binding pockets.22−24 The binding pattern of K-26 in human ACE is almost identical to that adopted by K-26 in the active site of Drosophila ACE (AnCE).25
Figure 2.
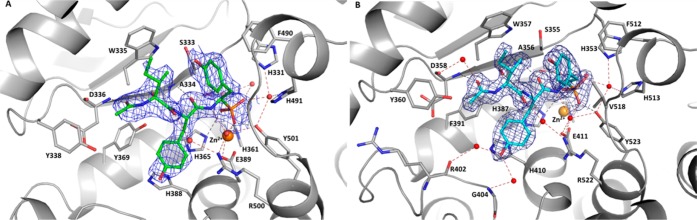
K-26 binding in both domains of ACE. (A) K-26 (green) bound in the N-domain of ACE. (B) K-26 (cyan) bound in the C-domain of ACE. Residues involved in K-26 interactions are shown as gray sticks. The zinc ion is shown as an orange sphere and water molecules in red. Potential hydrogen interactions and water mediated ones are presented with black and red dashed lines, respectively. Omit maps for K-26 bound to the N- and C-domains are shown at the 1σ contour level.
Figure 3.
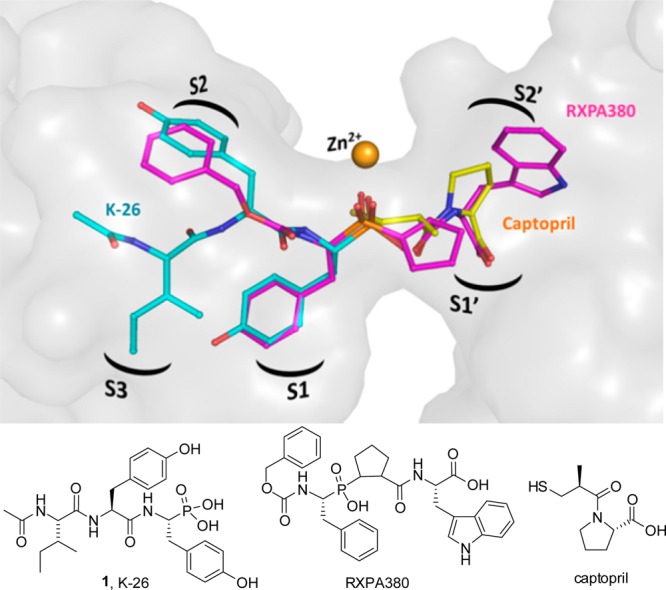
Binding motifs of K-26, captopril, and RXPA380 in the active site of ACE. K-26 is shown in cyan, captopril (PDB 1UZF) in green, and RXPA380 in pink (PDB 2OC2). Zinc is shown as an orange sphere. The binding pockets of the enzymes are labeled within the surface-rendered catalytic channel of C-domain ACE.
The shared activity of K-26 family metabolites is largely determined by the terminal nonproteinogenic amino acid (R)-AHEP, which has been demonstrated to be an essential component for potent ACE inhibitory activity. On its own, AHEP does not possess potent activity (IC50 > 1 μM); however, replacement of AHEP by tyrosine causes a 1500-fold decrease in ACE inhibitory activity.16 Alteration of stereochemistry also decreases the activity 10-fold. The crystal structure reveals the importance of the phosphonate for potent binding, as this is shown to coordinate with the zinc in the active site of the enzyme and form a hydrogen bond network with several conserved amino acids in both domains of ACE. The AHEP side chain fits into the S1 binding pocket, where the aromatic group of AHEP may form hydrophobic interactions with Val 518 (Thr 496 in N-ACE). Furthermore, the high resolution of the crystallographic data allowed for the identification of several key water molecules which may have a role in inhibitor binding. The AHEP seems to be stabilized at the active site by a network of water molecules anchoring K-26 at S1 in both crystal structures. Interestingly, additional water molecules interacting with K-26 were visible in the C-domain.
In the S2 binding pocket, the tyrosine side chain may form conserved aromatic stacking interactions with His 387 and His 410 (His 365 and His 388 in N-domain) and hydrophobic interactions with Phe 391 (Tyr 369 in N-ACE). Interestingly, synthesis of a phosphinic inhibitor library analogous to the inhibitor RXPA380 (Figure 3) showed that a Phe or Tyr in the S2 position of the inhibitor encouraged the most ACE binding potency in both domains, suggesting the importance of these interactions.24 Hydrogen bonds between the backbone of the tyrosine in K-26 and the backbone of Ala 356 (Ala 334 in N-ACE) are also evident. Additionally, the hydroxyl group of the tyrosine side chain makes water-mediated interactions with the backbone of Arg 402 and Gly 404 (Figure 2B). These two residues are conserved in both domains of ACE; however, Glu 403 (C-domain) is occupied by Arg 381 in the N-domain. The long polar side chain of Arg 381 may thus prevent these water-mediated interactions in the N-domain, decreasing the binding affinity for K-26.
The remaining structural features of the K-26 class of natural products are the third amino acid side chain and the N-terminal substitution, which occupies the S3 position. The N-terminal substitution has an effect on potency as shown with K-4 and 15-B-2, which have an N-terminal methyl group and inhibit somatic ACE with a 5–10-fold decrease in ACE inhibition, than the corresponding N-acetyl analogs. The origin of this activity difference likely derives from the ability of the N-acetyl carbonyl to form a hydrogen bond with the backbone nitrogen of Asp 358 (Asp 336 in N-domain) and the OH from Tyr 369 in the N-domain. These hydrogen bonding interactions with the N-acetyl tail are likely important interactions for the increased potency of the N-acetylated analogs in comparison to cationic N-methylated congeners.
Previous studies suggest that domain selectivity can be influenced by amino acids side chains occupying the S1 and S2 binding pockets of the enzyme.24,26,27 As K-26 is shown in the crystal structure to occupy the S1 and S2 pockets and a putative S3 binding pocket, this S3 binding pocket and area extending out further from the catalytic zinc is an unexplored area for inhibitor design. Synthetic analogs could focus on the S3 position where potential aromatic interactions with Trp 357 (Trp 335 in N-domain ACE), might provide enhanced potency. The crystal structures presented here have shown that the preferential C-domain selectivity observed with K-26 is provided by hydrophobic interactions at the S2 site along with water-mediated interactions with S2 and the N-acetyl. The nonconserved residues Glu 403 and Phe 391 (Arg 381 and Tyr 369 in N-domain ACE) seem again to be key positions to enhance domain selectivity.26 Although the domain selectivity of ACE inhibitors has been achieved through accessing peptidic inhibitors capable of occupying both the prime and nonprime binding pockets, the potential of a domain-specific inhibitor using only the nonprime binding sites has not been studied.
As K-26 family natural products are of microbial origin, and likely have not evolved to specifically inhibit human ACE, we endeavored to assay their interkingdom ACE-like activity. Biochemical characterization of several bacterial dicarboxypeptidases shows remarkable similarities in substrate and inhibitor preferences in comparison to mammalian ACE.28,29 In support of these observations, sequence alignment of several bacterial dicarboxypeptidases reveals a high degree of sequence homology between mammalian ACE and bacterial dicarboxypeptidases (Supporting Information Table 3 and Figure 1). This encouraged us to investigate the potential of the known potent mammalian ACE inhibitor, K-26, as an inhibitor of bacterial analogs of ACE. In order to explore the scope of interkingdom activity of K-26, we have recombinantly expressed, purified, and assayed a panel of ACE-like enzymes represented by phylogenetically distinct, bacterial zinc-dependent dicarboxypeptidases for inhibition by captopril and K-26 (Figure 4).
Figure 4.

Phylogenetic tree comparing ACE and related bacterial dicarboxypeptidases (DCP). Dicarboxypeptidases which have been overexpressed and purified in this study are shown in bold text. The optimal tree with the sum of branch length = 6.39523051 is shown.
Three bacterial enzymes were chosen: dicarboxypeptidases from the K-26 producing organism (K26DCP), from E. coli st. K-12 sbstr. MG1655 (EcDCP),28 and from Schwanella onedesis str. MR-1 (MR1DCP). K26DCP is the only putative zinc metallopeptidase present in the genome of the actinomycete Astrosporangium hypotensionis, the organism from which the tripeptide K-26 was originally isolated. MR1DCP was selected for the high similarity with human ACE, as it has a 43% sequence identity with both the N- and C- domains of human ACE. All three enzymes contain the characteristic HEXXH zinc-binding domain, characteristic of the zincin family of zinc metalloproteases. The dicarboxypeptidases EcDCP and the K-26 producer K26DCP both are classified specifically as a M3 zinc metalloproteases due to the presence of two additional glutamate residues, 30 and 37 amino acids C-terminal to the HEXXH zinc-binding domain that are believed to aid in coordinating the zinc in the active site of the enzyme.30 A glutamate residue present 29 residues downstream from the HEXXH zinc-binding domain, believed to be a ligand for zinc coordination in both MR1DCP and mammalian ACE, distinguishes these enzymes within the M2 subfamily of ACE zinc metalloproteases.
All three bacterial dicarboxypeptidases were found to efficiently hydrolyze the substrate FAPGG. Captopril was shown to be a potent, low nanomolar inhibitor of all the purified overexpressed dicarboxypeptidases (Table 2). However, K-26 was found to be a potent inhibitor of the mammalian ACE but a poor inhibitor of the bacterial ACE-like enzymes, including the dicarboxypeptidase encoded in the genome of the K-26 producer. Additionally, in previous studies, in the insect homologue of ACE, AnCE (Drosophila melanogaster), captopril has been shown to be a more potent inhibitor (Ki of 1.1 nM) of the enzyme than K-26 (Ki of 160 nM).25,31 Sequence alignment of human tACE with the most similar bacterial enzyme, MR1DCP, reveals five mutations in residues near the K-26 binding site (Supporting Information Figure 2). In the binding pocket, F391, G404, and F512 in tACE are mutated to Q396, S409, and Y515, altering the polarity of the pocket. However, the difference in potency between the mammalian and bacterial enzyme may arise from the mutation of F391 to Q396, which increases the net negative charge in the active site and likely deleteriously effects binding of our negatively charged substrate, K-26.
Table 2. Inhibition of Overexpressed Bacterial Dicarboxypeptidases by K-26.
| dicarboxypeptidase | type | captopril IC50 (M) | K-26 IC50 (M) |
|---|---|---|---|
| somatic ACE | M3 | 7.7 × 10–9 | 2.5 × 10–8 |
| K26DCP | M2 | 5.2 × 10–9 | 4 × 10–5 |
| EcDCP | M2 | 1.6 × 10–8 | 1.5 × 10–4 |
| MR1DCP | M3 | 9.2 × 10–9 | 1.1 × 10–5 |
In conclusion, we have illuminated the structure–activity relationships of the K-26 family of natural products in mammalian ACE, the enzyme target that was originally used in screening for their discovery from actinomycetes. These results provide structural insight that can be applied toward future development of potential domain-selective inhibitors through improving binding in the unexplored nonprime pockets of ACE. The inhibition data from bacterial ACE-like dicarboxypeptidases, together with previous data from Drosophila ACE, emphasize that, among all known groups of ACE-like dicarboxypeptidases, mammalian ACE seems uniquely inhibited by K-26. Indeed, the combined enzyme and substrate structural activity data suggest that the natural target for K-26 possesses an active-site architecture similar to mammalian ACE, prompting questions regarding potential interkingdom chemical ecological roles for the natural target of K-26. Future studies will investigate the potential for generation of improved domain selective analogs building from the AHEP pharmacophore into the unique binding pose of this family of natural products, and work to identify potential chemical ecological targets for AHEP functional natural products.
Acknowledgments
This study was supported by the University of Cape Town and the South African National Research Foundation to E.D.S. The crystallographic work reported here was supported by the Medical Research Council (U.K.) through a project grant (number G1001685) and a Wellcome Trust (U.K.) equipment grant (number 088464) to K.R.A. G.J.K. was supported in part by the D. Stanley and Ann T. Tarbell Endowment fund. Also, K.R.A. wishes to thank the scientists at PX station I03, Diamond Light Source, Didcot, Oxon (U.K.) for their support during X-ray diffraction data collection. B.O.B. and G.J.K were supported by National Institutes of Health Grant GM077189, the Vanderbilt Institute of Chemical Biology, and the Vanderbilt International Office. We thank Dr. Cody Goodwin for high resolution mass spectral data acquisition.
Glossary
Abbreviations
- AHEP
1-amino-2-(4-hydroxyphenyl)ethylphosphonic acid
- DCP
dicarboxypeptidase
- EcDCP
E. coli dicarboxypeptidase
- FAPGG
furylacryloyl-phenylalanyl-glycyl-glycine
- K26DCP
K-26 dicarboxypeptidase
- MR1DCP
Schwanella onedesis str. MR- 1 dicarboxypeptidase
- sACE
somatic angiotensin converting enzyme
- tACE
testicular angiotensin converting enzyme
- Z-FHL
Cbz-Phe-His-Leu-OH
Supporting Information Available
Synthetic details of the K-26 family of natural products, including NMR spectra for final products and synthetic intermediates, overexpression and purification of ACE constructs and bacterial dicarboxypeptidases, experimental methods for IC50 and Ki measurements, sequence homology and alignment for ACE-like enzymes of interest, primers used in the study, IC50 curves, and Dixon plots. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† (A.M.) Department of Biochemistry, University of Hyderabad, India 500046.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Newman D. J.; Cragg G. M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford J. M.; Clardy J. Bacterial symbionts and natural products. Chem. Commun. 2011, 47, 7559–7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohuchi I.; Kurihara K.; Shinohara A.; Takei T.; Yoshida J.; Amano S.; Miyadoh S.; Matsuhashi Y.; Shomure T.; Sezaki M. Studies on new angiotensin converting enzyme inhibitors, SF2513 A, B and C, Produced by Streptosporangium nondiastaticum. Sci. Rep. Meiji Seiki Kaisha 1988, 27, 46–54. [Google Scholar]
- Koguchi T.; Yamada K.; Yamato M.; Okachi R.; Nakayama K.; Kase H. K-4, a novel inhibitor of angiotensin I converting enzyme produced by Actinomadura spiculosospora. J. Antibiot. (Tokyo) 1986, 39, 364–371. [DOI] [PubMed] [Google Scholar]
- Yamato M.; Koguchi T.; Okachi R.; Yamada K.; Nakayama K.; Kase H.; Karasawa A.; Shuto K. K-26, a novel inhibitor of angiotensin I converting enzyme produced by an actinomycete K-26. J. Antibiot. (Tokyo) 1986, 39, 44–52. [DOI] [PubMed] [Google Scholar]
- Kido Y.; Hamakado T.; Anno M.; Miyagawa E.; Motoki Y.; Wakamiya T.; Shiba T. Isolation and characterization of I5B2, a new phosphorus containing inhibitor of angiotensin I converting enzyme produced by Actinomadura sp. J. Antibiot. (Tokyo) 1984, 37, 965–969. [DOI] [PubMed] [Google Scholar]
- World Health Organization. World Health Statistics. http://www.who.int/gho/publications/world_health_statistics/2012/en/ (accessed March 20, 2013).
- Morimoto T.; Gandhi T. K.; Fiskio J. M.; Seger A. C.; So J. W.; Cook E. F.; Fukui T.; Bates D. W. An evaluation of risk factors for adverse drug events associated with angiotensin-converting enzyme inhibitors. J. Eval. Clin. Pract. 2004, 10, 499–509. [DOI] [PubMed] [Google Scholar]
- Emanueli C.; Grady E. F.; Madeddu P.; Figini M.; Bunnett N. W.; Parisi D.; Regoli D.; Geppetti P. Acute ACE inhibition causes plasma extravasation in mice that is mediated by bradykinin and substance P. Hypertension 1998, 31, 1299–1304. [DOI] [PubMed] [Google Scholar]
- Nussberger J.; Cugno M.; Amstutz C.; Cicardi M.; Pellacani A.; Agostoni A. Plasma bradykinin in angio-oedema. Lancet 1998, 351, 1693–1697. [DOI] [PubMed] [Google Scholar]
- Wei L.; Alhenc-Gelas F.; Corvol P.; Clauser E. The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J. Biol. Chem. 1991, 266, 9002–9008. [PubMed] [Google Scholar]
- Soubrier F.; Alhenc-Gelas F.; Hubert C.; Allegrini J.; John M.; Tregear G.; Corvol P. Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 9386–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspard E.; Wei L.; Alhenc-Gelas F. Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J. Biol. Chem. 1993, 268, 9496–9503. [PubMed] [Google Scholar]
- Wei L.; Clauser E.; Alhenc-Gelas F.; Corvol P. The two homologous domains of human angiotensin I-converting enzyme interact differently with competitive inhibitors. J. Biol. Chem. 1992, 267, 13398–13405. [PubMed] [Google Scholar]
- Ehlers M. R.; Fox E. A.; Strydom D. J.; Riordan J. F. Molecular cloning of human testicular angiotensin-converting enzyme: the testis isozyme is identical to the C-terminal half of endothelial angiotensin-converting enzyme. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 7741–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntai I.; Bachmann B. O. Identification of ACE pharmacophore in the phosphonopeptide metabolite K-26. Bioorg. Med. Chem. Lett. 2008, 18, 3068–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntai I.; Phelan V. V.; Bachmann B. O. Phosphonopeptide K-26 biosynthetic intermediates in Astrosporangium hypotensionis. Chem. Commun. 2006, 4518–4520. [DOI] [PubMed] [Google Scholar]
- Holmquist B.; Bunning P.; Riordan J. F. A continuous spectrophotometric assay for angiotensin converting enzyme. Anal. Biochem. 1979, 95, 540–548. [DOI] [PubMed] [Google Scholar]
- Friedland J.; Silverstein E. A sensitive fluorimetric assay for serum angiotensin-converting enzyme. Am. J. Clin. Pathol. 1976, 66, 416–424. [DOI] [PubMed] [Google Scholar]
- Nchinda A. T.; Chibale K.; Redelinghuys P.; Sturrock E. D. Synthesis of novel keto-ACE analogues as domain-selective angiotensin I-converting enzyme inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4612–4615. [DOI] [PubMed] [Google Scholar]
- Schwager S. L.; Carmona A. K.; Sturrock E. D. A high-throughput fluorimetric assay for angiotensin I-converting enzyme. Nat. Protoc. 2006, 1, 1961–1964. [DOI] [PubMed] [Google Scholar]
- Natesh R.; Schwager S. L.; Evans H. R.; Sturrock E. D.; Acharya K. R. Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme. Biochemistry 2004, 43, 8718–8724. [DOI] [PubMed] [Google Scholar]
- Natesh R.; Schwager S. L.; Sturrock E. D.; Acharya K. R. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature 2003, 421, 551–554. [DOI] [PubMed] [Google Scholar]
- Corradi H. R.; Chitapi I.; Sewell B. T.; Georgiadis D.; Dive V.; Sturrock E. D.; Acharya K. R. The structure of testis angiotensin-converting enzyme in complex with the C domain-specific inhibitor RXPA380. Biochemistry 2007, 46, 5473–5478. [DOI] [PubMed] [Google Scholar]
- Akif M.; Ntai I.; Sturrock E. D.; Isaac R. E.; Bachmann B. O.; Acharya K. R. Crystal structure of a phosphonotripeptide K-26 in complex with angiotensin converting enzyme homologue (AnCE) from Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2010, 398, 532–6. [DOI] [PubMed] [Google Scholar]
- Anthony C. S.; Corradi H. R.; Schwager S. L.; Redelinghuys P.; Georgiadis D.; Dive V.; Acharya K. R.; Sturrock E. D. The N domain of human angiotensin-I-converting enzyme: the role of N-glycosylation and the crystal structure in complex with an N domain-specific phosphinic inhibitor, RXP407. J. Biol. Chem. 2010, 285, 35685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dive V.; Cotton J.; Yiotakis A.; Michaud A.; Vassiliou S.; Jiracek J.; Vazeux G.; Chauvet M. T.; Cuniasse P.; Corvol P. RXP 407, A phosphinic peptide, is a potent inhibitor of angiotensin I converting enzyme able to differentiate between its two active sites. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 4330–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha C. E.; Magliarelli Hde F.; Paschoalin T.; Nchinda A. T.; Lima J. C.; Juliano M. A.; Paiva P. B.; Sturrock E. D.; Travassos L. R.; Carmona A. K. Catalytic properties of recombinant dipeptidyl carboxypeptidase from Escherichia coli: a comparative study with angiotensin I-converting enzyme. Biol. Chem. 2009, 390, 931–940. [DOI] [PubMed] [Google Scholar]
- Riviere G.; Michaud A.; Corradi H. R.; Sturrock E. D.; Ravi Acharya K.; Cogez V.; Bohin J. P.; Vieau D.; Corvol P. Characterization of the first angiotensin-converting like enzyme in bacteria: Ancestor ACE is already active. Gene 2007, 399, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi S.; Shinoda S. Microbial metalloproteases and pathogenesis. Microbes Infect. 2000, 2, 91–98. [DOI] [PubMed] [Google Scholar]
- Williams T. A.; Michaud A.; Houard X.; Chauvet M. T.; Soubrier F.; Corvol P. Drosophila melanogaster angiotensin I-converting enzyme expressed in Pichia pastoris resembles the C domain of the mammalian homologue and does not require glycosylation for secretion and enzymic activity. Biochem. J. 1996, 318Pt 1125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.