

Abstract

The phosphatidylinositol 3-kinase (PI3K) signaling pathway plays important roles in cell proliferation, growth, and survival. Hyperactivated PI3K is frequently found in a wide variety of human cancers, validating it as a promising target for cancer therapy. We determined the crystal structure of the human PI3Kα–PI103 complex to unravel molecular interactions. Based on the structure, substitution at the R1 position of the phenol portion of PI103 was demonstrated to improve binding affinity via forming a new H-bond with Lys802 at the bottom of the ATP catalytic site. Interestingly, the crystal structure of the PI3Kα–9d complex revealed that the flexibility of Lys802 can also induce additional space at the catalytic site for further modification. Thus, these crystal structures provide a molecular basis for the strong and specific interactions and demonstrate the important role of Lys802 in the design of novel PI3Kα inhibitors.
Keywords: PI3K, PI103, crystal structure, drug design, cancer therapy
The lipid kinase family of phosphatidylinositol 3-kinases (PI3Ks) plays pivotal roles in many cellular processes, including proliferation, survival, differentiation, and metabolism.1−3 Class I PI3K, the best physiologically, biochemically, and structurally characterized member of the PI3K family, consists of four isoforms, α, β, γ, and δ. Each isoform is a heterodimer that comprises a p110 catalytic subunit and a p85 regulatory subunit. Upon insulin and growth factor stimulation, PI3Ks phosphorylate phosphatidylinositol-3,4-bisphosphate (PIP2) to produce phosphatidylinositol-3,4,5-triphosphate (PIP3). The cellular level of PIP3 is also tightly regulated by phosphatases, such as the phosphatase and tensin homologue (PTEN), which dephosphorylates PIP3 back to PIP2.4,5 The PI3K pathway is frequently deregulated in a wide range of tumor types as a result of hyperactivation of upstream growth factor signaling, mutation, or loss of PTEN,6 and oncogenic mutations in PIK3CA,7 which provides further evidence of the role of PI3K in tumorigenesis. Moreover, accumulating evidence indicates that hyperactivation of PI3Kα is inextricably linked to cancer survival and resistance to existing therapies in a great proportion of human cancers.8 Therefore, targeting PI3Ks with small-molecular-weight inhibitors provides an attractive opportunity for cancer therapy and for overcoming resistance to current therapies, and thus, significant efforts have recently been made to develop PI3K inhibitors.9
With multiple ongoing efforts in academic and industrial organizations to develop clinically relevant inhibitors against PI3K, a number of inhibitors have already entered clinical trials.2,10 PI103 is one of the first synthesized PI3K inhibitors; it belongs to the pyridinylfuranopyrimidine class and inhibits PI3K in an ATP-competitive manner with selectivity toward PI3Kα.11 PI103 has already demonstrated significant antitumor activity against several human tumor xenografts, especially those with well-established abnormalities in the PI3K pathway.12 Although PI103 failed to enter clinical trials, GDC0941, a drug candidate derived from PI103, is in phase I/II trials.7 Thus, PI103 provides a promising chemical template for the discovery and development of PI3K inhibitors. However, the precise interactions between PI103 and PI3Kα remain unknown. A better understanding of the interactions between PI103 and PI3K will facilitate the rational design of PI3K inhibitors with improved potency and selectivity.
To date, the solved PI3Kα–inhibitors crystal structures are extremely scarce and the nSH2 and iSH2 domains are not complete in only three of these structures (Supporting Information Table S1).13−15 To generate human wild-type full-length p110α and the nSH2 and iSH2 domains (hereafter termed “niSH2”) of p85α that would be suitable for crystallography, a fusion protein of the 6*his-TEV-p85α (318–615)-linker-p110α was expressed in the Bac-to-Bac Baculovirus expression system (Invitrogen) using a construct containing the iSH2 domain (467–568) of p85α fused at the N-terminus of the full-length p110α, as described previously.15,16 To produce a stoichiometric complex to enhance crystallization, analogous niSH2–p110α fusion constructs were designed, incorporating an additional linker between the iSH2 domain and the p110α in place of the thrombin cleavage site, which is prone to nonspecific proteolytic cleavage and accompanying heterogeneity. Using this method, a high purity of the 6*his-TEV-p85α (318–615)-linker-p110α homogeneous fusion protein was obtained via Ni-NTA, Sepharose Q, and gel filtration. The resulting complex contained all five p110α domains [an amino-terminal adaptor-binding domain (ABD), residues 1 to 108; a Ras-binding domain (RBD), residues 190 to 291; a C2 (protein-kinase-C homology-2) domain, residues 330 to 480; a helical domain, residues 525 to 696; and a carboxyl-terminal kinase domain, residues 697 to 1068], as well as the nSH2 (residues 318 to 430) and iSH2 (residues 431 to 615) domains of p85α (Figure 1). Through a sparse matrix screening of 1400 conditions and seeding optimization, a diffraction-quality crystal was produced for data collection, and an apo crystal was also incubated with 10 mM PI103. Diffraction data to a resolution of 2.6 Å were obtained for both the apo crystals and the PI103-containing ones and were refined to Rwork/Rfree values of 21.7/27.4 and 21.5/27.3, respectively (Supporting Information Table S2). Both structures maintained an overall triangle shape, with the long coiled-coil of iSH2 forming the base (Figure 1B and C).
Figure 1.
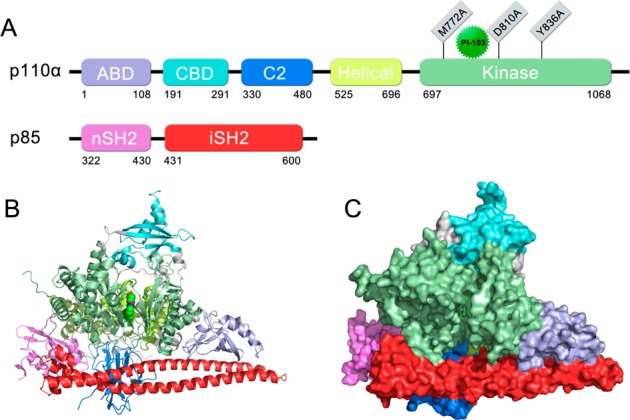
Overview of the p110α/niSH2 heterodimer. (A) Scheme of the domain organization. The same color coding is used throughout this figure. Gray regions are linkers between domains. Residue range for each domain is labeled under the domain diagram. (B) Diagram of the p110α/niSH2 heterodimer. The PI103 bound in the kinase domain is shown as spheres. (C) Surface diagram of the p110α/niSH2 heterodimer, alternate view. The PI103 bound in the kinase domain is shown as sticks.
The crystal structure of apo p110α revealed that the ATP-binding pocket is located in a cleft between the N- and C-terminal lobes of the kinase catalytic domain of p110α (Figure 2A), similar to the ATP binding sites in other protein kinases, with many of the enzyme/ATP interactions involving residues in the linker region between the two lobes. The crystal structure of p110α/p85α–PI103 appeared to verify that PI103 binds to the ATP-binding site on the p110α kinase domain (Figure 2B). PI103 adopts a flat conformation and sits between p110α residues Ile800, Asp810, Tyr836, Glu849, and Val851 on one side and residues Met922, Ile932, and Asp933 on the other side (Figure 2C). Three H-bonds are formed between PI103 and the active site residues of p110α. The morpholine oxygen donates an H-bond to the amide of the hinge Val851. The hydroxyl group of the phenol moiety makes two H-bonds to the carboxyl group of Asp810 and to the hydroxyl group of Tyr836.
Figure 2.

Binding pocket of p110α complexed with PI103. (A) Binding pocket of X-ray apo p110α (PDB ID: 4L1B); (B) Electron density map (2Fobs – Fcalc) of PI103 rendered at 1.0σ is shown in the binding pocket of the kinase domain; (C) X-ray complex of PI103 to p110α (PDB ID: 4L23). The structure of PI103 is shown as a stick representation, and the key binding site residues are shown as sticks. Hydrogen bonds between PI103 and the protein are shown as a dashed line; (D) Surface representation of PI103 bound p110α. Arrow indicates the direction of PI103 modification.
Upon closer inspection of the crystal structure of the p110α/p85α–PI103 complex, we noted that the distance between the phenol moiety of PI103 and the positively charged residue Lys802 is 3.96 Å, which may accommodate small substituted groups toward the side chain of Lys802 at the bottom of the ATP catalytic site for further modification to generate new PI103 derivatives with desirable potencies (Figure 2D). Based on this hypothesis, a series of substituted groups on the meta-site (R1 group in Table 1) of the phenol moiety in PI103 were introduced and probed the potential space (Supporting Information Scheme S1). As shown in Table 1, PI103 derivatives 9a, 9b, and 9c, in which a fluoro, chloro, and bromo group with different radius was replaced, respectively, for the original hydrogen atom in the R1 position of PI103, failed to enhance potency through the additional interactions with Lys802 (Figure 3A); based on the molecular modeling, we speculated that compound 9d, a slightly larger amino group with a positive charge in the R1 position, would cause a decreased potency due to the electrostatic repulsion between the incoming NH2 substitute and Lys802. Unexpectedly, compound 9d was as potent as PI103 against the PI3Kα (Figure 3B); substitution of the NH2 group in compound 9d with an OH group led to compound 9e, in which the oxygen atom acceptor was capable of forming an H-bond with Lys802. Moreover, molecular modeling using the crystal structure of the p110α/p85α–PI103 complex ascertained that the incoming OH group was hydrogen-bonded to the side chain of Lys802 (Figure 3C). Indeed, compound 9e was approximately 3-fold more potent against PI3Kα compared with PI103, with an IC50 value of 5.9 nM. Finally, large substituted groups on the R1 position of PI103 were further explored for the space. Replacing the hydrogen atom in the R1 position of PI103 with a nitro (compound 9f), methyl (compound 9g), and trifluoromethyl (compound 9h) group revealed that these compounds were negligible to inhibit the kinase activity of PI3Kα (Table 1). These results suggest that introducing a bulky group in the R1 position may produce dramatic steric clashes at the bottom of the ATP catalytic site.
Table 1. Effects of Test Compounds on the Kinase Activity of Class I PI3Kαa.
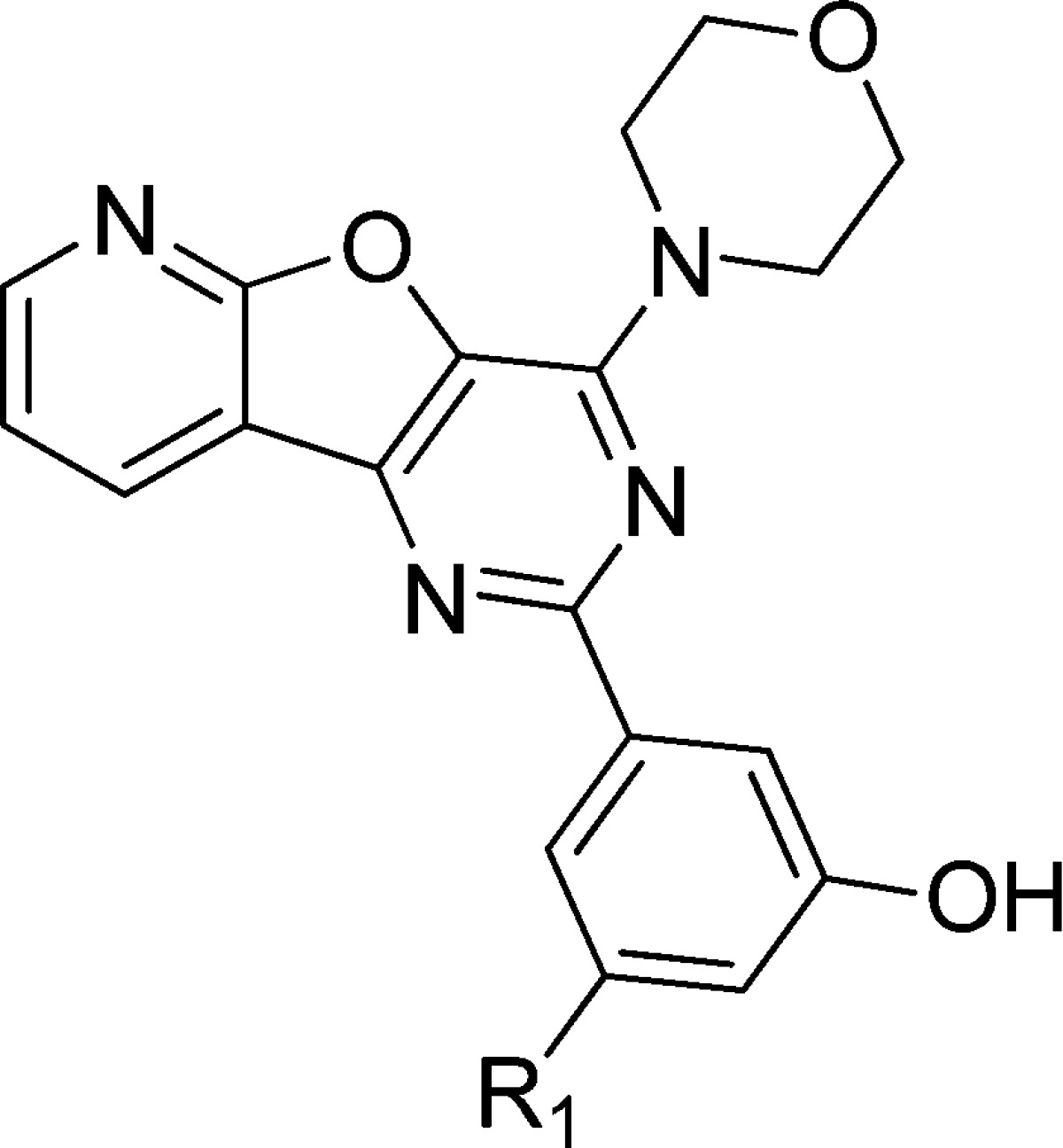
| cmpd | R1 | PI3KαIC50 (nM)b |
|---|---|---|
| PI103 | H | 17.9 ± 1.9 |
| 9a | F | 40.9 ± 5.8 |
| 9b | Cl | 990.0 ± 119.1 |
| 9c | Br | 1210.0 ± 246.8 |
| 9d | –NH2 | 20.6 ± 10.2 |
| 9e | –OH | 5.9 ± 0.4 |
| 9f | –NO2 | >10,000 |
| 9g | –CH3 | >10,000 |
| 9h | –CF3 | >10,000 |
Effects of test compounds on the kinase activity of class I PI3K were assessed as described in the Experimental Section.
IC50 values shown are the average of least three independent experiments in duplicate with typical variations of less than 20%.
Figure 3.

Binding modes of (A) compound 9a from docking; (B) compound 9d from X-ray (PDB ID: 4L2Y); (C) compound 9e from docking; (D) potential cavity of p110α induced by the binding of 9d. Key residues surrounding the active site of p110α are labeled, and carbon atoms are colored in pale green.
Structurally, Met772 exists in the flexible loop of PI3Kα, which is in an “up” conformation in the apo PI3Kα and a “down” conformation when PI103 is bound to the active site of PI3Kα. In the PI3Kα–PI103 complex, the hydroxyl group of PI103 forms bifurcated H-bonds with Asp810 and Tyr836 (Figure 2C), and Met772 interacts with PI103 via hydrophobic interactions (Supporting Information Figure S1). To investigate the effect of these residues on the activity of PI103 and its derivatives, M772A, D810A, and Y836A mutants of PI3Kα were generated, and the activity of PI103 and compound 9e against these mutants was determined. As shown in Table 2, compound 9e was more potent than PI103 against all three mutants, in particular the M772A and Y836A mutants; the fold change, defined by the ratio of the IC50 values of PI103 and compound 9e, was 3.0 for the wild type kinase and increased to 19.4 and 5.7 in the M772A and Y836A mutants, respectively (Table 2). These differences are most likely attributed to the extra hydrogen bonding interaction between the hydroxyl group in the R1 position of compound 9e and Lys802, which may stabilize the binding between compound 9e and the mutated PI3Kα.
Table 2. Effects of Compound 9e and PI103 on the Kinase Activities of Wild Type and Mutated PI3Kαa.
| cmpd (IC50, nM)b |
|||
|---|---|---|---|
| PI3Kα | PI103 | 9e | fold changec |
| wt | 17.9 | 5.9 | 3.0 |
| M772A | 1034.4 | 53.4 | 19.4 |
| D810A | 74.0 | 61.3 | 1.2 |
| Y836A | 796.3 | 139.8 | 5.7 |
Mutated PI3Kα were expressed and purified as described in the Experimental Section.
IC50 values shown are the average of at least three independent experiments performed in duplicate with typical variations of less than 20%.
The fold change was obtained from the ratio of the IC50 value of PI103 to that of compound 9e.
To determine the binding paradigms of PI103 derivatives to the p110α kinase catalytic domain, we incubated compounds 9a, 9d, and 9e with the p110α/p85α complex using a similar protocol for crystallization of the p110α/p85α–PI103 complex. After screening and optimization, a 2.8 Å resolution crystal structure of the human p110α/p85α in complex with compound 9d was obtained (Supporting Information Table S2). As shown in Figure 3B, generally, the interaction of compound 9d with the ATP catalytic site of p110α/p85α was similar to its interaction with the p110α/p85α–PI103 complex. However, the locally pronounced difference between the two crystal complexes was associated with the conformation of Lys802. The Lys802 side chain rotated away from the ATP catalytic site in the 110α/p85α–9d complex, whereas it pointed to the phenol moiety of PI103 in the ATP catalytic site in the 110α/p85α–PI103 complex (Supporting Information Figure S2). The induced conformation of the Lys802 side chain in the 110α/p85α–9d complex led to the vast space approaching the amino-substituted phenol moiety of 9d (Figure 3D), which can accommodate various large substituted groups in the cavity and provide a potential new direction to design more potent and selectivity inhibitors against PI3Kα based on compound 9d.
In summary, our PI3Kα–PI103 crystal complex demonstrated that the introduction of apt substitutes at the R1 position of the phenol moiety of PI103 results in derivatives with enhanced potencies, confirmed by compound 9e as a better analogue by forming a new H-bond with Lys802. Moreover, 9e was more tolerant to mutations of PI3Kα in the ATP catalytic site, such as M772A, D810A, and Y836A. Specially, the additional space near the phenol moiety was induced by a conformational change of Lys802 in the ATP catalytic site of the PI3Kα–9d crystal complex, which can be utilized for further design and optimization.
Acknowledgments
We thank Prof. Shaomeng Wang at the University of Michigan for fruitful discussions on the design of PI103 derivatives.
Glossary
Abbreviations
- PI3K
phosphatidylinositol 3-kinase
- ATP
adenosine triphosphate
- PIP2
phosphatidylinositol-3,4-bisphosphate
- PIP3
phosphatidylinositol-3,4,5-triphosphate
- PTEN
phosphatase and tensin homologue
- ABD
amino-terminal adaptor-binding domain
- RBD
Ras-binding domain
Supporting Information Available
Expression, purification, and crystallization of PI3Kα, synthetic procedures, assay details, and molecular modeling. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates for the apo PI3Kα, PI3Kα–PI103, and PI3Kα–9d structures have been deposited in the Protein Data Bank with access codes 4L1B, 4L23, and 4L2Y.
Author Contributions
‡ The manuscript was written through contributions of all authors. Y.Z., X.Z., Y.C., S.L., and Y.P. contributed equally.
This work was supported by the Program for New Century Excellent Talents in University (NCET-12-0355), the Shanghai Rising-Star Program (13QA1402300), the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” (2012ZX09301-001), the National Natural Science Foundation of China (81322046, 81302698, 81021062, 21002062, and 21102090), and the Knowledge Innovation Program of Chinese Academy of Sciences (KSCX2-EW-Q-3).
The authors declare no competing financial interest.
This paper published ASAP on December 11, 2013. The Supporting Information file was replaced and the revised version was reposted on December 16, 2013.
Supplementary Material
References
- Camps M.; Rückle T.; Ji H.; Ardissone V.; Rintelen F.; Shaw J.; Ferrandi C.; Chabert C.; Gillieron C.; Françon B.; Martin T.; Gretener D.; Perrin D.; Leroy D.; Vitte P. A.; Hirsch E.; Wymann M. P.; Cirillo R.; Schwarz M. K.; Rommel C. Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [DOI] [PubMed] [Google Scholar]
- Knight Z. A.; Gonzalez B.; Feldman M. E.; Zunder E. R.; Goldenberg D. D.; Williams O.; Loewith R.; Stokoe D.; Balla A.; Toth B.; Balla T.; Weiss W. A.; Williams R. L.; Shokat K. M. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell 2006, 125, 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q. W.; Knight Z. A.; Goldenberg D. D.; Yu W.; Mostov K. E.; Stokoe D.; Shokat K. M.; Weiss W. A. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellino R. C.; Muh C. R.; Durden D. L. PI-3 kinase-PTEN signaling node: an intercept point for the control of angiogenesis. Curr. Pharm. Des. 2009, 15, 380–388. [DOI] [PubMed] [Google Scholar]
- Foster J. G.; Blunt M. D.; Carter E.; Ward S. G. Inhibition of PI3K signaling spurs new therapeutic opportunities in inflammatory/autoimmune diseases and hematological malignancies. Pharmacol. Rev. 2012, 64, 1027–1054. [DOI] [PubMed] [Google Scholar]
- Davies M. A. The role of the PI3K-AKT pathway in melanoma. Cancer J. 2012, 18, 142–147. [DOI] [PubMed] [Google Scholar]
- Willems L.; Tamburini J.; Chapuis N.; Lacombe C.; Mayeux P.; Bouscary D. PI3K and mTOR signaling pathways in cancer: new data on targeted therapies. Curr. Oncol. Rep. 2012, 14, 129–138. [DOI] [PubMed] [Google Scholar]
- Sabbah D. A.; Brattain M. G.; Zhong H. Dual inhibitors of PI3K/mTOR or mTOR-selective inhibitors: which way shall we go?. Curr. Med. Chem. 2011, 18, 5528–5534. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Wang B. C.; Xiao Y. PI3K: a potential therapeutic target for cancer. J. Cell Physiol. 2011, 227, 2818–2821. [DOI] [PubMed] [Google Scholar]
- Denny W. A. Phosphoinositide 3-kinase α inhibitors: a patent review. Expert Opin. Ther. Pat. 2013, 23, 789–799. [DOI] [PubMed] [Google Scholar]
- Knight Z. A.; Chiang G. G.; Alaimo P. J.; Kenski D. M.; Ho C. B.; Coan K.; Abraham R. T.; Shokat K. M. Isoform-specific phosphoinositide-3 kinase inhibitors from an arylmorpholine scaffold. Bioorg. Med. Chem. 2004, 12, 4749–4759. [DOI] [PubMed] [Google Scholar]
- Fan Q.-W.; Cheng C.-K.; Nicolaides T. P.; Hackett C. S.; Knight Z. A.; Shokat K. M.; Weiss W. A. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007, 67, 7960–7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelker D.; Gabelli S. B.; Schmidt-Kittler O.; Zhu J.; Cheong I.; Huang C. H.; Kinzler K. W.; Vogelstein B.; Amzel L. M. A frequent kinase domain mutation that changes the interaction between PI3K and the membrane. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 16996–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon W. C.; Berndt A.; Williams R. L. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene 2012, 31, 3655–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacht M.; Qiao L.; Sheets M. P. St; Martin T.; Labenski M.; Mazdiyasni M.; Karp R.; Zhu Z.; Chaturvedi P.; Bhavsar D.; Niu D.; Westlin W.; Petter R. C.; Medikonda A. P.; Singh J. Discovery of a potent and isoform-selective targeted covalent inhibitor of the lipid kinase PI3Kα. J. Med. Chem. 2013, 56, 712–721. [DOI] [PubMed] [Google Scholar]
- Huang C. H.; Mandelker D.; Schmidt-Kittler O.; Samuels Y.; Velculescu V. E.; Kinzler K. W.; Vogelstein B.; Gabelli S. B.; Amzel L. M. The structure of a human p110α/p85α complex elucidates the effects of oncogenic PI3Kα mutations. Science 2007, 318, 1744–1748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.